Researchonline.lshtm.ac.uk

Taylor, MC; Kelly, JM (2006) pTcINDEX: a stable tetracycline-regulated expression vector for Trypanosoma cruzi. BMC Biotechnol,6. p. 32. ISSN 1472-6750 DOI: 10.1186/1472-6750-6-32
Downloaded from:
Please refer to usage guidelines at or alterna-tively contact
Available under license: http://creativecommons.org/licenses/by/2.5/
Research article
pTcINDEX: a stable tetracycline-regulated expression vector for
Trypanosoma cruzi
Martin C Taylor* and John M Kelly
Address: London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, UK
Email: Martin C Taylor* - [email protected]; John M Kelly - [email protected]
* Corresponding author
Published: 06 July 2006
Received: 23 February 2006Accepted: 06 July 2006
BMC Biotechnology 2006, 6:32
2006 Taylor and Kelly; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Licenswhich permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background: Trypanosoma cruzi is a protozoan pathogen of major medical importance in Latin
America. It is also an early diverging eukaryote that displays many unusual biochemical features. The
completion of the T. cruzi genome project has highlighted the need to extend the range of
techniques available to study gene function. To this end we report the development of a stable
tetracycline-dependent expression vector applicable to this parasite and describe in detail the
parameters of the system.
Results: We first produced T. cruzi cell lines that constitutively expressed bacteriophage T7 RNA
polymerase and the tetracycline repressor protein from a multicopy episome. An integrative vector
with an inducible expression site under the control of a tetracycline-regulatable T7 promoter
(pTcINDEX) was targeted to the transcriptionally silent ribosomal RNA spacer region of these
parasites and transformants selected using a T7 RNA polymerase-dependent hygromycin
resistance gene. To test the system we used two marker proteins, luciferase and red fluorescent
protein (RFP), and an endogenous parasite protein (a mitochondrial superoxide dismutase). In each
case we found that induction was both time and dose-dependent. Luciferase mRNA could be
induced by at least 100-fold, and luciferase activity up to 60-fold, within 24 hours of the addition of
tetracycline. When we examined RFP induction by confocal microscopy and fluorescence activated
cell sorter, we observed very high levels of expression (>1000-fold increase in fluorescence
intensity), although this was not synchronous throughout clonal populations. Induction of
superoxide dismutase resulted in an 18-fold increase in cellular activity. The observation that a
tagged version of the enzyme was correctly targeted to the mitochondrion demonstrates that our
expression system may also provide a high-throughput strategy for subcellular localisation.
Conclusion: Our results show that pTcINDEX represents a valuable addition to the genetic tools
available for T. cruzi. The vector system is sufficiently flexible that it should have widespread uses
including inducible expression of tagged proteins, generation of conditional knockout cell lines and
the application of dominant-negative approaches.
protozoa. This organism is the most important parasite in
Trypanosoma cruzi, the agent of Chagas disease, is a mem-
Latin America, while its close relatives Trypanosoma brucei
ber of the Kinetoplastidae, an early-diverging group of
and Leishmania cause African sleeping sickness and the
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
leishmaniases respectively. In addition to their medical
neered promoter in the absence of tetracycline. On
and veterinary significance, trypanosomes have been
addition of tetracycline, the repressor is released from the
studied as examples of primitive eukaryotes. They show
DNA and transcription is allowed to proceed. Initially, use
several biological peculiarities which have made them
was made of the T. brucei procyclin promoter [-
subjects of great interest. These include polycistronic tran-
ever, the system was found to be tightly regulated to a sim-
scription, trans-splicing of mRNA, mitochondrial RNA
ilar degree when a bacteriophage T7 promoter was utilised
editing, compartmentalisation of glycolysis and the utili-
This necessitated the integration of a T7 RNA
sation of a unique thiol, trypanothione, in place of glu-
polymerase gene into a transcriptionally active region of
tathione. Genome sequencing projects have recently been
the trypanosome genome prior to insertion of the con-
completed for each of the human pathogenic trypano-
struct containing the inducible gene. A similar regulatable
somatids, T. cruzi, T. brucei and Leishmania [ fully
expression system has now also been described for Leish-
exploit this vast amount of information it is essential that
mania based on an inducible copy of the endogenous
efforts are made to improve and extend the range of tools
ribosomal RNA (rRNA) pr
available for analysing the function of genes in vivo. Thisis particularly the case with T. cruzi, where technical limi-
In T. cruzi, inducible expression following transient trans-
tations currently restrict analysis of biological function.
fection with a plasmid has been reported . Morerecently a stable system has been reported by DaRocha et
The last few years have seen an explosion of new data on
al. [], in which the T7 polymerase and tetR genes were
gene function in T. brucei, largely due to the development
inserted into the tubulin gene array together with the
of regulated systems that allow inducible expression of
strong rRNA promoter. The effects, if any, of this promoter
both protein and double-stranded RNA [e sys-
on expression of endogenous genes flanking the insertion
tems can facilitate the study of gene function by over-
were not described, although a similar vector used in T.
expression [nditional knocko, or by RNA
brucei caused upregulation of genes downstream of the
interference (RNAi)-mediated down-regulation of gene
integrat. Detailed characterisation of this
expression RNAi is currently the method of
inducible cell line was not undertaken to assess the
choice for the analysis of gene function in T. brucei and
parameters of regulated expression. There have been no
can be used to inform studies on T. cruzi and Leishmania
further reports on its use or applications.
genes which have orthologues in T. brucei. However manytrypanosomatid genes are species-specific [. Since T.
Here we describe a stable tetracycline-inducible expres-
cruzi lacks the machinery for RNAi, specifically the AGO1
sion vector for T. cruzi that circumvents some of the
gene unpublished observations), approaches
potential problems associated with integration into an
such as gene deletion or expression of dominant-negative
endogenously transcribed locus. The system is based on
mutant proteins are of critical importance for studying
an integrative vector that facilitates inducible expression
function. However, both gene knockout and expression of
of specific genes in a transcriptionally quiescent locus and
mutant proteins can produce a lethal or deleterious phe-
engineered cell lines that constitutively express the T7
notype. It would therefore be advantageous to have a sys-
RNA polymerase and tetR genes from an episomal back-
tem that allows expression of transgenes in a controlled
ground. These experiments now provide a framework for
and repressible manner.
using stable inducible expression as a tool for studyinggene function in T. cruzi.
In general, trypanosomes do not appear to control expres-sion of protein coding genes at the level of transcription
initiation. The exceptions to this are the major surface
Production of cell lines stably expressing tetR and T7 RNA
glycoprotein genes of procyclic, metacyclic and blood-
stream forms of T. brucei ere RNA polymerase
Plasmid pLEW13, a construct designed to target the T. bru-
I (pol I)-dependent promoters can drive expression in a
cei β-tubulin locus, contains both T7 RNA polymerase and
developmental and locus specific manner. RNA polymer-
tetR genes with neo as a selectable mae
ase II (pol II)-dependent promoters for protein coding
electroporated T. cruzi CL-Brener epimastigotes with cir-
genes have not been unequivocally identified in trypano-
cular pLEW13 DNA (a gift from George Cross) and
somatids and there are no known examples of inducible
selected recombinant parasites on 200 µg ml-1 G418. Sta-
transcription units. Consequently, it has been necessary to
bly transformed parasites were obtained after six weeks,
import regulatable genetic machinery from other organ-
even though this vector contains no T. cruzi-derived
isms to create artificial inducible expression systems. Such
sequences. Southern analysis showed that the transform-
a system for T. brucei was first developed by Wirtz and
ants contained multiple copies of the input construct
Clayton ]. This relies on the bacterial tetracycline repres-
organised in a tandem array (data not shown). Circular
sor protein (tetR) to block transcription from an engi-
DNA in transformed T. cruzi usually replicates as an epi-
(page number not for citation purposes)





BMC Biotechnology 2006, 6:32
- 6-
56- T7 POL
AG -
cell lines stably expressing tetR and T7 RNA polymerase
Production of cell lines stably expressing tetR and T7 RNA polymerase. (A) Simplified map of pLEW13 indicating the
relative locations of the three transgenes [7]. (B) CHEFE analysis of chromosomal DNA isolated from CL-Brener [pLEW13]
epimastigotes showing aberrant migration of the pLEW13 DNA. Lanes 1–3, a 0.8% PFC agarose CHEFE gel (auto-algorithm set
for 300 kb-3 Mb separation). Lanes 1 (Saccharomyces cerevisiae size standards (Bio-Rad)) and 2 (CL-Brener [pLEW13]), the
ethidium bromide stained gel. Lane 3, an autoradiograph obtained with the T7 RNA polymerase probe. Lanes 4–6, a 1.0% PFC
agarose CHEFE gel (auto-algorithm set for 300 kb-1 Mb). Lanes 4 (S. cerevisiae size standards) and 5 (CL-Brener [pLEW13]),
the ethidium bromide stained gel. Lane 6 is an autoradiograph obtained with the T7 RNA polymerase probe. Molecular sizes
are given in kb. (C) Expression of the transgenes for T7 RNA polymerase and tetR in pLEW13 transformed epimastigotes. 10
µg total RNA was blotted and hybridised with either the T7 RNA polymerase (T7 POL) or tetR probes. (D) Splice acceptor sites used by T. cruzi to process the transcripts as mapped by RT-PCR. The AG dinucleotide sites of spliced leader addition identified following sequencing of the RT-PCR products are red and underlined. The numbers adjacent to the boxes indicate the distance in nucleotides between the sequence shown and the start codon of each gene. The T7 RNA polymerase is flanked by the T. brucei procyclin spliced leader acceptor site, whereas both neo and tetR are flanked by T. brucei actin spliced leader acceptor sites. In the case of the T7 RNA polymerase and tetR transcripts, only one addition site was identified; in the case of the neo transcript, three were found.
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
some of head-to-tail repeats of the input constr
RNA processing signals were being correctly utilised by T.
However, since the vector contained T. brucei β-tubulin
cruzi and that the T7 RNA polymerase and tetR mRNAs
coding sequences, which are very similar to the corre-
were therefore likely to be functional.
sponding T. cruzi gene (88% overall nucleotide identity,up to 96% in some regions), it was important to establish
Features of the tetracycline inducible expression vector
whether the pLEW13 tandem array was a circular episome
The inducible expression vector pTcINDEXas
or had resulted from multiple integrations into the tubu-
designed to integrate into the non-transcribed ribosomal
RNA spacer region upstream of the pol I-mediated tran-scription start siteethods). We targeted this region
Circular molecules show aberrant migration on pulsed
because, to our knowledge, it is the only section of the T.
field gels as their movement is independent of their
cruzi genome so far identified as being transcriptionally
molecular mass, in contrast to linear chromosomes. DNA
silent. In addition, the level of sequence conservation at
from CL-Brener epimastigotes transformed with pLEW13
this locus suggested that the construct could be targeted to
(CL-Brener [pLEW13]) was therefore subjected to con-
the corresponding region in multiple parasite strains. The
tour-clamped homogenous electric field gel electrophore-
targeting fragment is cloned as a Sac I cassette which can
sis analysis (CHEFE) under differing separation
be readily replaced to allow integration elsewhere in the
conditions (Fig). Using parameters designed to sepa-
rate the larger molecules (up to 3 Mb), the T7 RNApolymerase probe hybridised to a band of approximately
As a drug selectable marker we used the hygromycin B
2 Mb and to a smear of higher molecular weight material
phosphotransferase (hyg) gene under the control of a non-
(Flane 3). When the DNA was fractionated under
repressible T7 promoter, thus converting antibiotic resist-
conditions optimal for separation of molecules of
ance into a digenic trait. In pLEW13 transformed cells that
between 300 kb and 1 Mb, the hybridising band ran at
constitutively express the T7 RNA polymerase, this
680 kb, again accompanied by a smear of apparently
arrangement serves a second function. In the presence of
higher molecular weight materine 6). The
hygromycin, the requirement for T7 RNA polymerase to
migration of the pLEW13 construct is therefore independ-
drive expression of hyg removes the necessity for the con-
ent of its molecular weight indicative of a circular episome
tinued use of G418 to maintain the pLEW13 construct
containing multiple copies of the T7 RNA polymerase and
and selects for trypanosomes with active T7 RNA polymer-
tetR genes. Southern analysis of genomic DNA also indi-
ase. The inducible expression cassette in the pTcINDEX
cated no linkage between the T. cruzi α-tubulin genes and
vector contains a tetracycline-dependent T7 promoter,
the T7 RNA polymerase (data not shown).
with the tet operator sequence (tetO, cCTATCAgTGAT-AGa, where upper case indicates bases important in tetR
To check expression of the transfected genes, RNA was
binding) placed immediately downstream. The multiple
prepared and analysed by northern blotting. This showed
cloning site is flanked at its 3'-end by the intergenic
that both T7 RNA polymerase and the tetR gene were
sequence from the T. cruzi actin locus to provide a polya-
expressed at high levels (Fig. ypanosomes each
denylation signal, and at the 5'-end by the splice acceptor
mRNA is processed by trans-splicing which results in the
site from the ribosomal protein P2β locus. Sequences
addition of a 5' spliced leader sequence of 39 nucleotides
from this region have been shown to enhance the expres-
the RNA processing signals in pLEW13 were
sion of transfected genes [Finally, we incorporated a
derived from T. brucei it was necessary to establish that the
T7 RNA polymerase transcription terminator into the con-
transgenic mRNAs were correctly spliced. For each gene,
struct to block run-through transcription of sequences
primers were designed to sequences approximately 150–
downstream of the integration site.
250 bp into the ORF and used in conjunction with aprimer to the T. cruzi spliced leader in an RT-PCR reaction
To test the capability of the vector to mediate tetracycline-
(Methods). The resulting products were cloned and
regulatable expression we cloned the genes encoding fire-
sequenced. Each splice addition site could be mapped to
fly luciferase (Luc) and red fluorescent protein (RFP) into
an AG dinucleotide located downstream of a polypyrimi-
the multiple cloning ods). Spe I linearised
dine tract (Fig case of the T. brucei actin inter-
forms of the resulting constructs (pTcINDEX-Luc and
genic sequence upstream of neo, three separate splice sites
pTcINDEX-RFP) were then used to transform CL-Brener
were identified, all of which were upstream of the start
[pLEW13] epimastigotes that constitutively express T7
codon. Only one of these was identified in the tetR mRNA
RNA polymerase and tetR. Integration into the rRNA locus
which has the same flanking sequence. In the case of the
(illustrate) was confirmed by Southern analy-
procyclin splice acceptor site upstream of the T7 RNA
sis. This showed linkage of both of the transgenes to the
polymerase, only the site previously mapped in T. brucei
endogenous 18S rRNA gene, (for examp
was utilised. This analysis indicated that the T. brucei
The appearance of novel fragments in the lanes containing
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
II R-NTS/P
STUFFER GATATC gag caa aag ctc att tct gaa gag gac ttg
The T. cruzi
inducible expression vectors pTcINDEX and pTcINDEX-C-myc
The T. cruzi inducible expression vectors pTcINDEX and pTcINDEX-C-myc. (A) Map of pTcINDEX. The grey box
adjacent to the multiple cloning site (MCS) indicates the ribosomal protein P2β splice acceptor site [25]. The hatched box indi-
cates the T. cruzi actin intergenic region. The black box (T) is the T7 transcriptional terminator. Black flags represent T7 pro-
moters and the oval identifies the location of the tet operator. R-NTS/P is the ribosomal non-transcribed spacer and promoter
region used to target the construct. Roman numerals I and II indicate the two halves of the targeting sequence which are
cloned in the opposite order to their position in the genome (see Fig. 3A). The white flag indicates the location of the pol I
transcription start site [24]. Following insertion of a gene of interest into the MCS, the construct can be linearised with Spe I
(dotted line) to facilitate targeting to the rRNA non-transcribed spacer region. The vector is built on a pUC19 backbone (not
illustrated for clarity) and confers ampicillin resistance on E. coli. The sequence of the MCS is shown above the map indicating
useful restriction sites. Nae I and Nru I were incorporated as blunt end sites to facilitate cloning of genes which contain the
other restriction sites. (B) Map of pTcINDEX-C-myc. The inducible cassette alone is shown for clarity. The rest of the vector
is identical to pTcINDEX. Features are as shown in A, except that the BPP1-myc fusion gene has been inserted into the Bam HI/
Nru I sites of pTcINDEX (Methods). The white box labelled "stuffer" indicates the dispensable BPP1 ORF [48]. This can be
removed by digestion with one of the MCS enzymes and Eco RV and replaced with the gene of interest. The c-myc epitope tag
is indicated by a green box. The Eco RV cleavage site and the translated sequence of the c-myc tag (underlined) are indicated to
allow easy design of in-frame fusions with the epitope tag. Note the Nru I site is absent in this plasmid.
DNA from the transformants (9.5 kb with the luciferase
as we wished to see if the gene returned to a repressed
and 18S rRNA prob
state. Northern analysis was performed (Figand the
the 18S rRNA and RFP prob, lanes 6 and 8)),
relative level of luciferase RNA measured at each time
which were absent from the CL-Brener [pLEW13] lanes,
point using a phosphorimager. In the lane containing
were diagnostic of targeted integration into the non-tran-
RNA from non-treated cells, the signal detected was not
scribed spacer region upstream of the 18S rRNA gene.
significantly above the background measured from anirrelevant piece of the membrane. This indicates a tightly
Induction of luciferase in pTcINDEX-Luc transformants
regulated system with a very low level of "leaky" transcrip-
We first investigated the induction of luciferase RNA in a
tion. 24 hours after the addition of tetracycline, the level
polyclonal line of pTcINDEX-Luc transformed cells. Tetra-
of luciferase mRNA was found to have increased dramati-
cycline was added once to epimastigotes in early mid-log-
cally (F). The mRNA levels at later time points
arithmic growth phase (approximately 106 parasites ml-1)
declined gradually. The change in luciferase RNA levels
and aliquots removed every 24 hours for RNA purifica-
was mirrored in the level of luciferase activity ).
tion. No further tetracycline was added during this period,
The enzyme level increased considerably over 24 hours
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
I II R-NTS/P
Kpn I
Kpn I
Nco I 1.6 kb
Nco I
Nco I
1 2 3 4 5 6 7 8
Integration of pTc
INDEX-Luc and pTcINDEX-RFP into the ribosomal non- transcribed spacer
Integration of pTcINDEX-Luc and pTcINDEX-RFP into the ribosomal non- transcribed spacer. (A) Configura-
tion of correctly targeted constructs showing relevant restriction sites. R-NTS/P represents the ribosomal non-transcribed
spacer/promoter region with the white flag indicating the promoter [24]. The targeting fragment is designed to integrate
upstream of the rRNA transcription start. The dotted line represents the position of the Spe I site introduced into the spacer
to facilitate linearization. This site is absent from the genomic DNA. The crossed lines indicate the sites of homologous recom-
bination. The double headed arrow shows the region of the 18S rRNA gene used as a probe when assessing integration. The
other symbols are as in Fig. 2. The configurations for integration of pTcINDEX-Luc and pTcINDEX-RFP are shown. The
expected fragment size following a targeted integration is illustrated below each map. (B) Southern analysis of the pTcINDEX-
Luc and pTcINDEX-RFP transformants Arrowheads indicate fragments specific to the transformants following hybridisation
with the 18S rRNA probe. These bands also hybridise specifically to the full-length luciferase or RFP probes. Lanes 1,3,5,7 con-
tain DNA from CL-Brener [pLEW13], lanes 2 and 4 from CL-Brener:pTcINDEX-Luc [pLEW13]. Lanes 6 and 8 contain DNA
from CL-Brener:pTcINDEX-RFP [pLEW13]. DNA in lanes 1–4 was digested with Kpn I and in lanes 5–8 with Nco I. The probes
used are indicated below each autoradiograph. A second smaller band (1.6 kb) which hybridises to the 5' end of the RFP probe
(lane 8) migrated off the bottom of this gel. Molecular sizes are shown in kb.
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
and continued to increase up to 48 hours. Thereafter it
U 1 2 3 4 5 6 7 Days
declined gradually. When the cells were washed after 24hours exposure to tetracycline and resuspended in tetracy-
cline-free medium, the luciferase activity reached a peak at48 hours but had declined almost to background levelsthree days later (Fed line).
To examine the relationship between tetracycline concen-tration and the induction of luciferase activity, a culture of
pTcINDEX-Luc transformed CL-Brener [pLEW13] epimas-
tigotes was divided into 10 individual flasks and tetracy-
cline added at a range of concentrations (FigC). The
cells were incubated for 24 hours, then harvested and the
luciferase activity measured (Methods). There was negligi-
ble increase in luciferase activity over the level in non-
treated cells at concentrations up to and including 1 ng
ml-1. The increased activity became significant following
treatment with 5 ng ml-1 and continued to increase with
concentration before levelling off at 500 ng ml-1
Time (hrs)
At tetracycline concentrations of 1 or 2 µg ml-1, the extent
of induction decreased approximately two-fold, an effect
that was reproducible. At higher levels of tetracycline there
were detectable increases in parasite doubling time. The
optimal increase of luciferase activity that was achieved
was approximately 60-fold over background. This is less
than the increased level of the corresponding transcript
nce of control mech-
anisms at the level of translation or instability of the luci-
ferase protein in this context.
It has previously been noted that different clones trans-
formed with the same tetracycline-regulated construct in
[tetracycline] (µg ml-1)
T. brucei will exhibit differences in both the backgroundlevel and the extent of inducible expression of the trans-
Induction of luciferase by tetracycline
fected ge. This variability has been ascribed, in
Induction of luciferase by tetracycline. (A) Expression
part, to epigenetic factors operating differentially on each
of luciferase mRNA in a polyclonal line of tetracycline
site of integrationxamine whether this variation
treated cells. pTcINDEX-Luc transformed epimastigotes
occurred in T. cruzi, we isolated several clones from inde-
were harvested each day following the addition of tetracy-
pendent transfections. Variability was indeed observed
cline to the cultures (5 µg ml-1). Lane U is the uninduced cell
(Table ground level of luciferase activity var-
line. Lanes 1–7 represent days following induction. 1.25 µg
ied from 700 to 5000 relative light units (RLU) per 5 × 104
total RNA was loaded in each lane. The blot was hybridised with a 1.7 kb Bam HI/Sal I fragment containing the luciferase
cells. The background remained constant in a given clone
ORF. (B) Luciferase activity over a time course. Cells were
over time. The level of induction after 24 hours varied
induced and aliquots removed for enzyme assay at indicated
from 2 to 37-fold between different clones in this experi-
timepoints. Dashed line: cells were induced for 24 hours
ment. In T. cruzi all the ribosomal RNA arrays are present
then washed twice in tetracycline-free medium and resus-
at one chromosomal locus, in contrast to the situation in
pended in tetracycline-free medium. Solid line: cells were
T. brucei. However the sequence across this locus is una-
induced with tetracycline then incubated with no further
vailable and it has not yet been possible to determine if
treatment. (C) Effect of tetracycline concentration on the
there is a relationship between the level of expression and
level of luciferase activity. 10 identical flasks of epimastigotes
the site of integration.
(as above) were treated with different concentrations of tet-racycline for 24 hours. Cells were harvested and the luci-
Cell-by-cell examination of the induction process using
ferase activity measured (Methods). The Y axis indicates the fold increase in luciferase activity above uninduced cells, nor-
malised, to the amount of protein present in each extract.
To determine how individual cells responded to tetracy-cline, we examined clones isolated following transforma-
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
Table 1: Inducible luciferase activity in independent clones
(Fig. FACS analysis showed that in the first 24 hours
transformed with pTcINDEX-Luc
post-induction, there was a significant shift in the fluores-cence profile, with 35% of cells showing a 10–1000 fold
Cell line
0.5 µg ml-1 tet
increase in intens line). The profile shifted
rightwards over time but did not sharpen, indicating that
fluorescence intensity varied between individual cells,
confirming the observation made by microsco
The maximal shift was seen on day 5 (Fig. e),
when 14 % of the cells were found to exhibit a 1000–10000 fold increase in fluorescence. Even at this stage
Each clone was induced with 0.5 µg ml-1 tetracycline. After 24 hours,
however, 34% of cells remained in the 0–10 arbitrary flu-
extracts were tested for luciferase activity (Methods). Each extract was assayed in triplicate. Controls were identical cultures maintained
orescence units (AFU) range.
in the absence of tetracycline. The activity is represented as mean relative light units per 5 × 104 epimastigotes. Figures in parentheses
We also examined the extent of variation in both back-
represent standard error of the mean.
ground and inducibility in the RFP expressing clones.
tion with pTcINDEX-RFP (Methods). Epimastigotes of
Again we observed a ra. For exam-
clone CL:RFP C2 were maintained in tetracycline-free
ple, with clone CL:RFP B1 there was only slight induction,
medium, then an aliquot was fixed onto a slide. Tetracy-
whereas all the other clones showed significant levels. In
cline was added to the remainder of the culture. An aliq-
this experiment, tetracycline was added every three days to
uot of cells was removed and fixed onto a slide every 24
maintain the level of induction.
hours for eight days. It was apparent that RFP expressionhad been highly induced by the third day, since the cell
Addition of an rRNA promoter to pLEW13 results in higher
pellet had a red tinge visible to the eye. The cells were
background expression levels
stained for DNA and examined by confocal microscopy.
In an attempt to produce a more homogeneous induction
No red fluorescence was visible in the uninduced popula-
profile, we constructed a derivative of pLEW13 in which
ti). After 24 hours a few cells displayed faint
the T7 RNA polymerase and tetR genes were transcribed
fluorescence, and after three days some cells were
from the T. cruzi rRNA promoter (Methods). Cells were
extremely bright. The number of visibly fluorescent cells
transformed with this plasmid (pTcrRNA-T7tet) and
increased over time. After eight days the majority of cells
selected at 100 µg ml-1 G418. The transformants were
exhibited some red fluorescenchough there
resistant to 2 mg ml-1 G418, with no lag phase, indicative
was variation in the level. This suggested that induction,
that the rRNA promoter was driving high level expression
as measured using this parameter, does not occur at the
of the neo gene. These cells were then electroporated with
same rate or to the same degree in all cells of a given clonal
the inducible vector pTcINDEX-RFP. Parasites were
cloned immediately after electroporation. FACS analysisof several independent clones confirmed that expression
To examine this variation further we quantified the distri-
of RFP was tetracycline-regulated, but again the response
bution of induced fluorescence in the population by FACS
was heterogeneous within clonal populations).
an experiment the tetracycline treat-ment was carried out at 0.5 µg ml-1, as this concentration
As these clones showed a somewhat higher background
was optimal for expression, at least in the case of luciferase
level of RFP expression (especially clone CL [pTcrRNA-T7Tet]:RFP C6), we tested the luciferase construct in this
Table 2: Inducible RFP expression in independent clones
background. 5 clones were generated in the CL [pLEW13]
transformed with pTcINDEX-RFP.
line and 5 in the CL [pTcrRNA-T7Tet] background. Each
Cell line
0.5 µg ml-1 tet
cell line was induced for 48 hours with 250 ng ml-1 tetra-cycline, and then assayed for luciferase activity. The results
indicated inducible luciferase activity in all cell
lines tested. However, there was a much lower level of
leakiness in the CL [pLEW13] cell line than the CL
[pTcrRNA-T7Tet] cell line. Whilst the former exhibited a
10–30 fold increase in luciferase activity, the latter dis-
played only a 5 to 9-fold increase. A similar effect was
Each clone was induced with 0.5 µg ml-1 tetracycline. After 5 days,
noted in the inducible system created for Leishmania ].
cells were fixed and analysed on a FacsCalibur. Controls were
Consequently, for applications in which a tightly regu-
identical cultures maintained in the absence of tetracycline. The data
lated system is required, the CL [pLEW13] cell line
are presented as percentage of cells registering greater than 6
appears to be much more suitable.
arbitrary fluorescence units (AFU).
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
A UNINDUCED
B INDUCED
in cloned cells by microscopy
RFP expression in cloned cells by microscopy. Fluorescence microscopy of cloned pTcINDEX-RFP transformed epimas-
tigotes before and after induction with tetracycline (5 µg ml-1). An aliquot was removed, and fixed every 24 hours for eight
days. Cells were examined on a Zeiss LSM 510 microscope. Panel A shows the uninduced population and panel B the same cul-
ture 8 days after induction. In each case 1: DNA stained with TOTO-3 (green), 2: RFP expression (red), 3: phase image and 4
is a merged image.
Expression and localisation of epitope-tagged superoxide
Two clones were characterised (A1 and A2). With both, an
induced band was visible after western blotting (Fig. ).
To assign a biological role to a protein it is necessary to
In the induced cells, the corresponding bands were visible
know its subcellular location. Generation of specific anti-
on a Coomassie stained gel ). The upregulated
bodies against every protein of interest is costly, time-con-
SOD was enzymatically active (Fig. induced
suming and not always successful. We therefore made a
cells showing a 14- and 18-fold increase over the control
derivative of pTcINDEX with a c-myc epitope tag inserted
lines, respectively. This represents the total cellular SOD
next to the polylinker to facilitate localisation of induced
activity. Since there are four distinct isoforms in trypano-
proteins (pTcINDEX-C-myc, Fig. ). This could also pro-
somatidsit is clear that the level of SOD A over-
vide a simple method to follow the induction by western
expression considerably exceeds 18-fold.
blotting. To test the vector, we chose the T. cruzi superox-ide dismutase A gene (TcSOD A), which encodes an iso-
It was important to confirm that the SOD A was targeted
form with a predicted mitochondrial targeting sequence.
correctly since overexpression might lead to mis-targeting
The T. brucei orthologue of this gene is targeted to the
or blocking of the trafficking pathway. Cells were stained
with an antibody against the carboxyl-terminal epitopetag and examined by microsco. The immun-
The TcSOD A gene was inserted into pTcINDEX-C-myc
ofluorescence showed targeting of the induced protein to
such that the epitope tag was located at the carboxyl-ter-
the single lattice-like mitochondrion of the trypanosome
minus of the fusion protein ). CL-Brener
with a concentration in a rod-like structure next to, or on
[pLEW13] epimastigotes were transformed as previously.
top of, the kinetoplast (mitochondrial) DNA. The exact
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
made up of multiple head-to-tail copies of the vector. Thisorganisation has commonly been observed for episomal
constructs in both Leishmania and T. cruzi is
thought to arise by insertional duplication. We used a
multicopy episome, rather than single integrated copies of
120 HOURS
the T7 RNA polymerase and tetR genes, to decrease thepossibility of selection for mutations which could rescuedominant-negative or conditional knockout cell lines.
Such rescue mutants occur readily in the T. brucei systemwhich relies on single copies of each gene,]. Epi-somes have been shown to be maintained in the absenceof selection for up to six months, and during passagethrough mammalian cells and insect vectors in T. cruzi[.
The inducible expression vector (pTcINDEX) wasdesigned to integrate into the transcriptionally silentribosomal RNA spacer region. We judged this to be impor-
Figure 6 lysis of expression of RFP in a cloned cell line
tant for two reasons. Firstly, so that in its repressed state,
FACS analysis of expression of RFP in a cloned cell
with the tetR protein bound tightly to the tetO, the inte-
line. Tetracycline (0.5 µg ml-1) was added and samples were
grated expression cassette did not block the transcription
removed at specific timepoints. The level of fluorescence in
of downstream genes, and secondly, so that run-through
the population was measured by counting 5 × 103 cells per
pol II transcription occurring from upstream genes did
timepoint on a FACScan. The filled curve is the uninduced
not interfere with repression. In trypanosomatids, linked
population. The traces for induced cells are shown as over-
protein coding genes are organised into large polycis-
laid lines: blue, 24 hours: red, 48 hours and green, 120 hours
tronic transcription units and transcriptional termination
post-induction. The Y axis indicates the number of cells
has not been fully characterised for any RNA polymerase.
counted, whilst the X axis shows the level of RFP expression
The pTcINDEX vector was also designed so that the hyg
drug selectable marker was under the control of a consti-tutive T7 promoter. Thus expression of T7 RNA polymer-ase is necessary for cells to display resistance tohygromycin and continued selection with G418 is no
nature of this structure is unclear, but the consistent prox-
longer required to maintain the presence of the pLEW13
imity to the kinetoplast suggests a possible role in protec-
tion of the replicating kDNA from reactive oxygen species.
In the pLEW13 transformed cells, our experiments indi-
cate that the level of inducible expression may vary from
We have constructed a stable tetracycline-regulated
gene to gene, but that any background, due to insuffi-
expression vector for T. cruzi and tested several of the asso-
ciently tight repression of promoter activity, is likely to be
ciated features using two marker genes, luciferase and
low. With pTcINDEX-RFP transformed cells, we were una-
RFP, and an endogenous gene TcSOD A. These experi-
ble to detect any fluorescence by microscopy, and only a
ments demonstrate that the system should be sufficiently
low level by FACS, in the absence of tetracycline (0.2% –
robust to have widespread application in the functional
3% of cells counted depending on the clonfilled
analysis of parasite genes. Initially, we produced stable
pTcINDEX-Luc transformed
cell lines that constitutively expressed T7 RNA polymerase
cells, detection of the luciferase transcript on northern
and the tetR protein using a vector system (pLEW13) orig-
blots was tetracycline-dependent (Fig. r,
inally constructed for the African trypanosome. We were
there was a reproducibly detectable level of enzyme activ-
able to confirm constitutive expression of both genes in
ity associated with non-induced cells. This background
transformed cells, even though the input plasmid com-
appeared relatively constant in a given clone, although it
pletely lacked T. cruzi sequences. This type of phenome-
did vary between clones. We also noticed that the level to
non has previously been observed in Leishmania [
which luciferase activity was induced (up to 60-fold) was
but not in T. cruzi . Addition of the spliced leader sequence
low compared to that of the mRNA (>100-fold), suggest-
to each transcript occurred at the same sites as used in T.
ing that expression may be regulated at the level of trans-
brucei (Fig. nalysis of the transformed cells indi-
lation or that the luciferase protein may be less stable in
cated that the pLEW13 was propagated as an episome
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
RFP in individual cells, even within a cloned population,ranging from <10- to >1000-fold. When the machinery
Cl [pTcrRNA-T7tet]:RFP A5
required for inducible expression (the T7 RNA polymeraseand tetR genes) was placed under the control of an rRNApromoter, the induction profile did not become morehomogeneous, but the background expression increasedsignificantly. Thus, no advantage was gained and repres-sion was decreased using this construct. This could be dueto high level expression of tetR resulting in aggregationand loss of function as has been postulated in the Leishma-nia system [.
Cell-by-cell analysis in the manner described here, hasnot, to our knowledge, been performed with the T. bruceior Leishmania inducible expression systems, although a
Cl [pTcrRNA-T7tet]:RFP C6
heterogeneous pattern of induction has been observedusing a tetracycline-regulated promoter to drive expres-sion of GFP in yeastis type of variation is a com-mon feature of eukaryotic cells and is thought to reflectthe inherently stochastic nature of gene expression at thelevel of both transcription and transl-twork from several laboratories has shown that stochasticelements play a significant part in generating "noise" ingene expression, i.e. the variation in expression of a givenprotein between genetically identical individuals in a pop-ulation under the same conditions []. Thus, the pat-tern of RFP fluorescence observed using FACS analysisand microscopy may be regarded as a "snapshot" of thefluctuating levels of RFP expression that occur even within
Cl [pTcrRNA-T7tet]:RFP D1
a clonal population. Indeed, a recent re hasshown that even when a marker gene (GFP) is integratedinto the T. cruzi genome under the control of a strong con-stitutive rRNA promoter, the FACS profile of a stablytransformed cell line is remarkably heterogeneous, withapproximately 25–30% of cells expressing little or nodetectable GFP at a given time. This suggests that variationin protein levels between individual cells may be an inher-ent feature of T. cruzi gene expression, rather than a con-sequence of episomal expression of the polymerase andrepressor genes. It is notable that the T. cruzi genome con-tains many highly polymorphic multigene familiesencoding surface proteins which are co-expressed in a
FACscan analysis
clones in the CL [pTcrRNA on of RFP in 3 indepe
-T7tetR] background ndent
given population. Therefore, stochastic expression of sur-
FACscan analysis of expression of RFP in 3 independ-
face antigens between individual cells of a population
ent clones in the CL [pTcrRNA-T7tetR] background.
may be an important immune evasion strate. It has
Tetracycline (0.5 µg ml-1) was added and samples were
been hypothesised that micro-organisms benefit from
removed and fixed after 6 days. The level of fluorescence in
noise in gene expression as this allows a population to
the population was measured by counting 5 × 103 cells per
respond more rapidly to changes in their environment
timepoint. The filled area indicates the uninduced cells, while the green line represents the induced population. The Y axis
and decreases the chances of cells becoming mired in
indicates the number of cells counted, whilst the X axis
inappropriate epigenetic st].
shows the level of fluorescence in AFU.
The availability of our inducible expression system willprovide new approaches for the functional analysis of
Both microscopy and FACS analysis showed that there
genes in T. cruzi. It will allow the study of proteins that
was a wide variation in the level of inducible expression of
may be toxic if constitutively expressed, enable the gener-
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
Table 3: Inducible luciferase activity in independent clones transformed with pTcINDEX-Luc in pLEW13 or pTcrRNA-T7Tet
background.
Cell line
0.25 µg ml-1 tet
CL-L1B6 (pTcrRNA-T7Tet)
CL-L1C4 (pTcrRNA-T7Tet)
CL-L2A4 (pTcrRNA-T7Tet)
CL-L2D4 (pTcrRNA-T7Tet)
CL-L1A6 (pTcrRNA-T7Tet)
Each clone was induced with 0.25 µg ml-1 tetracycline. After 48 hours, extracts were tested for luciferase activity (Methods). Each extract was assayed in triplicate. Controls were identical cultures maintained in the absence of tetracycline. The activity is represented as mean relative light units per 5 × 104 epimastigotes. Figures in parentheses represent standard error of the mean.
ation of conditional knockouts of essential genes and
negative mutants of T. cruzi, an organism for which RNAi
facilitate functional knockouts by means of overexpres-
based approaches are not applicable.
sion of dominant-negative protein mutants. The level ofoverexpression achieved with SOD A suggests that
a dominant-negative approach will be feasible, since in
Parasite maintenance and genetic manipulation
such a system the mutated protein must be expressed at
Epimastigotes of T. cruzi CL-Brener were maintained at
significantly higher levels than the endogenous enzyme.
27°C in RPMI-1640 medium as described previously], except that we used 5% tetracycline-free fetal calf
Modulation of expression levels by changing the concen-
serum (Autogen Bioclear). Parasites were transformed by
tration of tetracycline could also be important for condi-
electroporation using a Bio-Rad Gene Pulser II, placed
tional knockout experiments. This will enable the
into fresh medium and incubated for 48 hours to allow
transfected gene to be expressed at a similar level to the
expression of the drug-selectable marker. The appropriate
endogenous copy, thereby preventing unforeseen pheno-
drug was then added (G418 at 100–200 µg ml-1 or hygro-
typic consequences due to overexpression. An advantage
mycin at 100 µg ml-1) and the cells incubated for a further
of using tetracycline as the inducer is that the expression
four to six weeks to allow selection of transformants. For
system can be applied to the study of enzyme function
direct cloning, parasites were resuspended in 24 ml of
throughout the life-cycle. For example, it should be possi-
fresh medium directly after electroporation. 1 ml was then
ble to investigate the development of transformed para-
transferred to each well of a 24-well plate and the cells
sites within tissue-culture cells using the tetracycline
allowed to grow for 48 hours prior to addition of the
analogue doxycycline, which has been used to regulate
selective drug. Typically, between 2 and 5 clones were gen-
murine gene expression in transgenic (Tet-O
erated per 24-well plate.
The combination of episomally expressed T7 RNApolymerase and tetR with an inducible vector which can
integrate into the rRNA locus in both group I and group II
The inducible expression vector pTcINDwas
parasites also means that this system is transferable to any
based on the T. brucei RNAi vector pZ(a gift from
strain of T. cruzi. pTcINDEX and pTcINDEX-C-myc are
Paul Englund). First, the inverted promoter fragment of
freely available to members of the trypanosomatid
pZJM, which contains bi-directional T7 promoters, was
research community.
isolated by digestion with Kpn I and Bam HI. This frag-ment was then subcloned into pGEM3zf+ (Promega) to
produce vector pGEMT7Tet2. In parallel, a hyg gene
We have designed and tested a user-friendly tetracycline-
flanked by the processing signals from the T. cruzi glyco-
regulatable expression vector, pTcINDEX, for the proto-
somal glyceraldehdye-3-phosphate dehydrogenase
zoan parasite T. cruzi. This vector has been used to gener-
gene[] was inserted into the Eco RV site of pBlue-
ate cell lines bearing inducible copies of luciferase, RFP
script KS(-). A constitutive T7 promoter was then inserted
and SOD A. The levels of repression and induction
upstream of the hyg gene after generation of the appropri-
achieved lead us to believe that this vector will be useful
ate fragment by PCR. This 2.4 kb cassette was isolated fol-
for creating both conditional knockouts and dominant-
lowing Kpn I and Sac I digestion and sub-cloned into
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
Inducible expression of
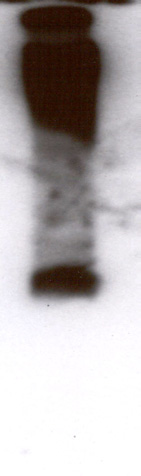
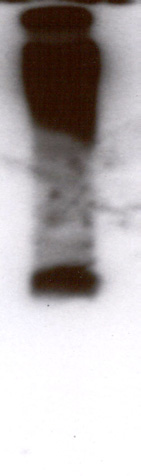
Inducible expression of TcSOD A. Two clones containing pTcINDEX-SOD A9E10 on a CL-Brener [pLEW13] background
were induced with 0.25 µg ml-1 tetracycline for five days. Replicate cultures were grown in the absence of tetracycline. Protein
extracts were made and analysed by SDS-PAGE, western blotting and enzyme assay. A) Western blot of a gel stained with the
mouse monoclonal 9E10. A single band of approximately 25 kDa was recognised by the antibody. B) Coomassie stained SDS-
PAGE gel showing lysates from control and induced populations of clones A1 and A2. Note the intense band appearing in the
induced lanes, at the position of the band recognised by the antibody in A. C) Relative SOD activities of the trypanosome
lysates as measured using the SOD 525 assay system. Each assay was performed in triplicate. Clone A1 showed a 14:1 ratio of
SOD activity between induced and uninduced cells, while clone A2 showed an 18:1 ratio.
pGEMT7Tet2, upstream of the fragment derived from
two pieces of 1.8 kb and 0.3 kb to allow the introduction
pZJM, to create pGEMhygT7-3. Since the pGEM backbone
of a unique Spe I site for vector linearization prior to trans-
contains an additional unwanted T7 promoter, the whole
fection. These pieces were generated using the primer
insert fragment was liberated by Bam HI/Sac I digestion
and inserted into pUC19∆H (pUC19 with the Hind III sitedeleted by end-filling).
The ribosomal RNA non-transcribed spacer and promoter
GTGTCTAGTACATC, 5'-
region were amplified from genomic DNA of T. cruzi in
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
Immunofluorescence lo
calisation of epitope tagged TcSOD A
Immunofluorescence localisation of epitope tagged TcSOD A. Cells were induced as in Fig [8]. The parasites were
fixed in paraformaldehyde and stained with mouse monoclonal anti-c-myc 9E10. Slides were examined on a Zeiss LSM 510 con-
focal microscope. The epitope tagged protein is shown as green fluorescence with the DNA stained red. Arrows indicate the
strong staining of a structure adjacent to the kinetoplast (K). The nucleus is indicated (N). The white bar indicates 5 µm. The
phase image is shown for comparison. Both images show cells in the process of dividing.
The fragments were ligated together then cloned into the
into Xho I/Hind III digested
Sac I site immediately downstream of the hyg cassette (as
shown in Fig. 2) to produce plasmid pTcIRi. To constructthe inducible expression vector, we modified pTcIRi by
The splice acceptor site from the ribosomal protein P2β
removing the antisense promoter and adding a polylinker
locus was amplified from pTREX-n [ (a gift from Mari-
and RNA processing signals. Briefly, pTcIRi was partially
ano Levin), using primers which added an Xho I site to the
digested with Sac I and Hind III. The Sac I/Hind III frag-
5' end and a Not I site to the 3'end. This 212 bp fragment
ment containing the hyg gene and the sense strand induc-
was cloned into the corresponding sites of the polylinker.
ible promoter was cloned into pGEM3zf+ (Promega) to
The actin intergenic sequence [Ah
create pGEMTcI. The T7 transcriptional terminator from
contains a putative polyadenylation signal, was amplified
pTcIRi was amplified with primers 5'-
from genomic DNA of T. cruzi using primers: 5'-CCCG-
GATCGTCGCGAGGCAGGCCCAAGCA and 5'-CCCGA-
GGATCCTCGCGAATCAGGTGCTAGCCCGCT and 5'-
TATCGTCAGACATCCTTAGAA. The resulting 424 bp
TTTAAGCTTGATCCCCGGATATAGTT. This fragment,
fragment was then digested with Bam HI and Eco RV and
which also incorporated a multiple cloning site (under-
cloned into the Bam HI and Nru I sites of the polylinker.
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
The entire insert was again transferred to pUC19 via a par-
in-frame with the carboxyl terminal epitope tag under the
tial Sac I/Hind III digest to produce the final expression
control of the inducible T7 promoter. The construct was
confirmed by DNA sequencing.
The luciferase coding sequence was obtained from pGEM-
Nucleic acid analysis
luc (Promega). The plasmid was digested with Sal I and
DNA and RNA were prepared and purified using Qiagen
end-filled by the Klenow fragment. The gene was then
kits as per manufacturer's instructions. RNA was quanti-
excised by digestion with Bam HI. pTcINDEX was digested
fied using a 2100 Bioanalyzer with RNA 6000 Nano
with Bam HI and Nru I and the luciferase gene was ligated
Labchip (Agilent). Southern and northern blotting were
into the vector to produce pTcINDEX-Luc. To obtain the
carried out using standard protocols. Reverse transcriptase
gene encoding the red fluorescent protein (RFP), con-
PCR (RT-PCR) was carried out using the Access RT-PCR kit
struct pTEX-Red [as first digested with Bam HI and
(Promega) and primers:
Bgl II and re-ligated to delete awkward restriction sites.
The modified plasmid was then cut with Spe I and the
Spliced Leader sense 5'-GGGGGATCCACAGTTTCTGTAC-
ends filled by Klenow treatment. The RFP gene was liber-
ated by digesting the linear plasmid with Cla I and clonedinto Nae I/Cla I sites of pTcINDEX to create pTcINDEX-
T7 Polymerase antisense 5'-TCGTAAGACTCATGCTCAA
Neo antisense 5'-CCTCGTCCTGACAGTTCAT
pLEW13 was modified to include a T. cruzi rRNA pro-moter to drive expression of the T7 RNA polymerase, neo
tetR antisense 5'-TGCCTATCTAACATCTCA
and tetR genes. Briefly the Sac I fragment carrying therRNA promoter and upstream spacer region was removed
The products were cloned into pGEM-T (Promega) and
from pTcINDEX and subcloned into pUC19 to make
sequenced using a dye terminator cycle-sequencing kit
pUC-TcrRNA. The T7 RNA polymerase, neo and TetR genes
(Applied Biosystems) and an ABI Prism 377 DNA
were removed from pLEW13 on a 5.9 kb fragment using
sequencer. For CHEFE analysis parasite blocks were made
EcoRV and Stu I. This fragment was then cloned into the
as de. The chromosomes were resolved on a
unique Spe I site in the rRNA promoter fragment of pUC-
CHEFmapper system (Bio-Rad) using the auto-algorithm
TcrRNA, such that the transcription initiation point was
and conditions as detailed in figure legends.
upstream of the T7 polymerase gene. This derivative ofpLEW13 was named pTcrRNA-T7Tet.
Induction of gene expression
For induction experiments, epimastigotes were cultured
We created an epitope tagging vector by cloning the BPP1-
in 25 cm3 flasks at 27°C and maintained in logarithmic
myc fusion gene from pTEX-BPP1-9E10 into Bam HI/Nru
growth phase (106 – 107 cells ml-1). Control cells were
I digested pTcINDEX [. This fusion gene contains a
grown in tetracycline-free medium, whilst the induced
unique Eco RV site between the BPP1 ORF and the c-myc
cells were cultured in medium supplemented with the
tag such that any gene of interest can replace the BPP1
stated concentration of tetracycline. We found that the
coding sequence and be cloned in-frame with the tag (Fig.
doubling time of wild type parasites was unaffected by
epitope tag encodes the sequence EQKLISEEDL*,
low concentrations of tetracycline, but was increased by
where * indicates a translational stop. This vector was
13% at 5 µg ml-1 and by 30% at 10 µg ml-1. Inductions
named pTcINDEX-C-myc where the uppercase C denotes
were carried out over variable time courses as stated in fig-
that the tag is fused to the carboxyl terminal of the protein
ure legends.
of interest. To make an inducible tagged copy of TcSOD A,the gene (>Tc00.1047053509775.40) was amplified
from genomic DNA of the CL-Brener strain using the fol-
Epimastigotes transformed with pTcINDEX-Luc were
grown as described above. At each time point an aliquotwas removed, pelleted and washed in PBS (137 mM NaCl,
SOD A F: gggggatccATGTTGAGACGTGCGGTGAA
4 mM Na2HPO4, 1.7 mM KH2PO4, 2.7 mM KCl). Cell pel-lets were frozen in liquid nitrogen and stored at -80°C.
SOD A R: ggttgatatcTTTTATTGCCTGCGCAT
For the luciferase assay, the pellet was resuspended in 500
µl of cell culture lysis reagent (Promega). Lysates were vor-
where underlining indicates restriction sites introduced
texed for 15 seconds and the debris removed by centrifu-
for ease of cloning. The 699 bp product was digested with
gation. Activity was measured using the luciferase assay
Bam HI and Eco RV and ligated into Bam HI/Eco RV
system (Promega) and light emission measured on a β-
digested pTcINDEX-C-myc, such that the SOD A ORF was
plate counter (Wallac). The linear detection limits of the
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
counter were measured using serial dilutions of Quanti-
involved in this study. JMK participated in the conception
Lum recombinant luciferase (Promega). Protein concen-
and design of the study and helped to draft the manu-
trations were determined by the BCA assay (Pierce) using
script. Both authors read and approved the final manu-
equivalent amounts of cells lysed in PBS, as the lysis rea-
gent is incompatible with the protein assay.
Fluorescence microscopy and FACS analysis of RFP
We would like to thank George Cross, Paul Englund and Mariano Levin for
the kind gifts of plasmids pLEW13, pZJM and pTREX-n respectively. We
RFP expression was examined by confocal microscopy on
would also like to acknowledge the assistance of Nick Dorrell, Sara Prickett
a Zeiss LSM 510 Axioplan microscope. Transformed para-
and Chandrabala Shah in using the Agilent 2100 bioanalyzer, FacScan and FacsCalibur respectively. Shane Wilkinson provided invaluable assistance
sites were induced as described above. At each time point,
with the SOD assays. This work was funded by the Wellcome Trust.
an aliquot of cells (107) was removed, pelleted, washed inPBS and then fixed for 30 minutes in 4% paraformalde-
hyde/PBS. Cells were then washed and resuspended in 5
El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G,
ml PBS. 20 µl of the suspension was dotted onto a single
Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westen-
well of a 12-well slide. DNA was stained by adding 50 nM
berger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A,Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P,
TOTO-3 (Molecular Probes) in 10 mg ml-1 RNAse A/0.1%
Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Sil-
saponin/PBS to each well, incubating at room tempera-
veira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T,Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E,
ture for 20 minutes, then washing twice in PBS. Slides
Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T,
were mounted in 1:1 PBS/glycerol. For FACS analysis,
Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nel-
cells were fixed as above and finally resuspended at 107
son S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Petters-son U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez
parasites ml-1. 5 × 103 – 104 cells per time point were
DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tar-
counted on a FacScan or FacsCalibur (Becton Dickinson).
leton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B,Wortman J, White O, Fraser CM, Stuart KD, Andersson B:
Data were analysed using Cellquest™ software (BD Sci-
Science 2005, 309:409-415.
Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bar-tholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U,
Protein extraction and analysis
Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Als-
For western blot and SOD activity assays, cells were pel-
mark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K,
leted, and washed once in PBS. The cells were pelleted
Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN,Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum
again and resuspended in lysis buffer (PBS supplemented
T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR,
with proteinase inhibitors, Roche). The cell suspension
Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, JonesK, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V,
was freeze-thawed three times in liquid nitrogen then son-
Line A, Lord A, Macleod A, Mooney PJ, Moule S, Martin DM, Morgan
icated. Membrane debris was removed by centrifugation
GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS,
(10,000 g for 10 mins). The supernatant was removed to
Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C,Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ,
a sterile tube and stored at -80°C. SDS-PAGE and western
Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wan-
blotting were carried out as per standard protocols. The
less D, Wang S, White B, White O, Whitehead S, Woodward J,Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb
western blots were probed with mouse monoclonal c-Myc
AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM,
(9E10) (cat no. sc-40, Santa Cruz Biotechnology Inc.)
Melville SE, El-Sayed NM:
diluted 1:2000. For SOD activity assays the Bioxytech™
Science 2005, 309:416-422.
Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman
SOD 525 (Oxis Research) kit was used as per manufac-
M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou
Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G,Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clay-ton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J,
Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser
To check the localisation of the tagged SOD A, epimastig-
A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C,Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N,
otes were fixed in 4% paraformaldehyde and dried onto
Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S,
slides. The slides were stained with mouse monoclonal c-
Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H,
Myc (9E10) (diluted 1:200) and then Alexafluor 488 con-
Oliver K, O'Neil S, Pentony M, Pohl TM, Price C, Purnelle B, QuailMA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Rob-
jugated goat anti-mouse (diluted 1:400 Molecular
ertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz
Probes). DNA was stained with DAPI. Slides were exam-
DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares
ined on a Zeiss LSM 510 confocal laser scanning micro-
S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H,Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stu-
art KD, Barrell B, Myler PJ: Science 2005, 309:436-442.
Wirtz E, Clayton C:
Science 1995,
MCT designed the vectors and all derivatives thereof
except where stated, and carried out all practical work
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
Wirtz E, Hartmann C, Clayton C:
Martinez-Calvillo S, Hernandez R:
Gene 1994,
Nucleic Acids Res 1994, 22:3887-3894.
Wirtz E, Hoek M, Cross GA:
Vazquez MP, Levin MJ:
Nucleic Acids Res 1998, 26:4626-4634.
Gene 1999,
Wirtz E, Leal S, Ochatt C, Cross GA
Alsford S, Kawahara T, Glover L, Horn D:
chem Parasitol 1999, 99:89-101.
Mol Biochem
Wang Z, Morris JC, Drew ME, Englund PT
Parasitol 2005, 144:142-148.
Wilkinson SR, Prathalingam SR, Taylor MC, Ahmed A, Horn D, Kelly
J Biol Chem
Free Radic
LaCount DJ, Bruse S, Hill KL, Donelson JE:
Biol Med 2006, 40:198-209.
Dufernez F, Yernaux C, Gerbod D, Noel C, Chauvenet M, Wintjens
Mol Biochem Parasitol 2000, 111:67-76.
R, Edgcomb VP, Capron M, Opperdoes FR, Viscogliosi
Ulbert S, Cross M, Boorstein RJ, Teebor GW, Borst P:
Nucleic Acids Res 2002, 30:3919-3926.
Radic Biol Med 2006, 40:210-225.
Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel
Papadopoulou B, Roy G, Ouellett
Mol Biochem
Parasitol 1994, 65:39-49.
Mol Microbiol 2000, 35:542-552.
Schlecker T, Schmidt A, Dirdjaja N, Voncken F, Clayton C, Krauth-
Drozdz M, Palazzo SS, Salavati R, O'Rear J, Clayton C, Stuart K
Embo J 2002, 21:1791-1799.
J Biol Chem 2005, 280:14385-14394.
El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal
Ariyanayagam MR, Oza SL, Guther ML, Fairlamb
G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Pea-
cock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark
Biochem J 2005, 391:425-432.
UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton
Gaunt MW, Yeo M, Frame IA, Stothard JR, Carrasco HJ, Taylor MC,
JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM,
Mena SS, Veazey P, Miles GA, Acosta N, de Arias AR, Miles
Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D,
Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger
Nature 2003, 421:936-939.
S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall
Blake WJ, M KAE, Cantor CR, Collins JJ
Nature 2003, 422:633-637.
Science 2005, 309:404-409.
McAdams HH, Arkin A:
Ullu E, Tschudi C, Chakraborty T:
Proc Natl Acad Sci U S A 1997, 94:814-819.
Cell Microbiol 2004, 6:509-519.
Norris AJ, Stirland JA, McFerran DW, Seymour ZC, Spiller DG, Lou-
DaRocha WD, Otsu K, Teixeira SM, Donelson JE:
don AS, White MR, Davis JR
Mol Endocrinol 2003, 17:193-202.
Mol Biochem Parasitol 2004, 133:175-186.
Elowitz MB, Levine AJ, Siggia ED, Swain PS:
Laufer G, Schaaf G, Bollgonn S, Gunzl A:
Science 2002, 297:1183-1186.
J Biosci 2005, 30:21-30.
Mol Cell Biol 1999,
Raser JM, O'Shea EK:
Science 2004, 304:1811-1814.
Lee MG, Van der Ploeg LH:
Raser JM, O'Shea EK:
Annu Rev Micro-
Science 2005, 309:2010-2013.
biol 1997, 51:463-489.
Pedraza JM, van Oudenaarden A:
Yan S, Myler PJ, Stuart K:
Science 2005, 307:1965-1969.
Mol Biochem Parasitol 2001,
Rosenfeld N, Young JW, Alon U, Swain PS, Elowit
Science 2005, 307:1962-1965.
Wen LM, Xu P, Benegal G, Carvaho MR, Butler DR, Buck GA
DaRocha WD, Silva RA, Bartholomeu DC, Pires SF, Freitas JM,
Macedo AM, Vazquez MP, Levin MJ, Teixeira SM:
Exp Parasitol 2001, 97:196-204.
Dhir V, Allen CL, Field MC:
Parasitol Res 2004, 92:113-120.
Buscaglia CA, Campo VA, Frasch AC, Di Noia JM
Exp Parasitol 2005,
Microbiol 2006, 4:229-236.
Kelly JM, Ward HM, Miles MA, Kendall G:
Kaern M, Elston TC, Blake WJ, Collins JJ:
Nat Rev Genet 2005,
Nucleic Acids Res 1992,
Xie W, Chow LT, Paterson AJ, Chin E, Kudlow JE:
Murphy WJ, Watkins KP, Agabian
Cell 1986, 47:517-525.
Oncogene 1999, 18:3593-3607.
Rudenko G, Le Blancq S, Smith J, Lee MG, Rattray A, Van der Ploeg
Kendall G, Wilderspin AF, Ashall F, Miles MA, Kelly JM:
Embo J 1990, 9:2751-2758.
Mol Cell Biol 1990,
Wilkinson SR, Meyer DJ, Taylor MC, Bromley EV, Miles MA, Kelly JM
(page number not for citation purposes)
BMC Biotechnology 2006, 6:32
J Biol Chem 2002, 277:17062-17071.
Bromley EV, Taylor MC, Wilkinson SR, Kelly JM Int J Parasitol 2004, 34:63-71.
Bishop RP, Miles MA: Mol Biochem Parasitol 1987, 24:263-272.
scientist can read your work free of charge
"BioMed Central will be the most significant development for
disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community
peer reviewed and published immediately upon acceptance
cited in PubMed and archived on PubMed Central
yours — you keep the copyright
Submit your manuscript here:
(page number not for citation purposes)
Source: https://researchonline.lshtm.ac.uk/11537/1/1472-6750-6-32.pdf
B activ (rv)drs176 020712 final rd.indd
B Complex with Benfotiamine and Quatrefolic® » Supports Carbohydrate Metabolism* » Supports Healthy Nervous System/Adrenal/Immune Function* Neurologic & Cognitive » Supports Cardiovascular Health* » Supports Healthy Mental Function and Mood* B Activ™ contains the entire spectrum of B vitamins to support adrenal and neurological functions. It features activated forms
Microsoft word - galanterkrishnan25.doc
GalanterKrishnan25 4/6/2004 12:19 PM "Bread for the Poor": Access to Justice and the Rights of the Needy in India† Marc Galanter* & Jayanth K. Krishnan** India is rightly acclaimed for achieving a flourishing constitutional order, presided over by an inventive and activist judiciary, aided by a proficient bar, supported by the state and cherished by the public. At the same time, the courts and tribunals where ordinary Indians might go for remedy and protection are beset with massive problems of delay, cost, and ineffectiveness. Potential users avoid the courts; in spite of a long-standing reputation for litigiousness, existing evidence suggests that Indi-ans avail themselves of the courts at a low rate and the rate seems to be falling.1 Still, the courts remain gridlocked.2 There is wide agreement that