Poitoutlab.ca


This article appeared in a journal published by Elsevier. The attached
copy is furnished to the author for internal non-commercial research
and education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or
licensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of the
article (e.g. in Word or Tex form) to their personal website or
institutional repository. Authors requiring further information
regarding Elsevier's archiving and manuscript policies are
encouraged to visit:


Author's personal copy
Biochimica et Biophysica Acta 1801 (2010) 289–298
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
Glucolipotoxicity of the pancreatic beta cell
Vincent Poitout a,b,c,⁎, Julie Amyot a,b, Meriem Semache a,b, Bader Zarrouki a,b, Derek Hagman a,Ghislaine Fontés a
a Montreal Diabetes Research Center, CRCHUM, University of Montreal, Montreal, QC, Canadab Department of Medicine, University of Montreal, Montreal, QC, Canadac Department of Biochemistry, University of Montreal, Montreal, QC, Canada
The concept of glucolipotoxicity refers to the combined, deleterious effects of elevated glucose and fatty acid
Received 2 July 2009
levels on pancreatic beta-cell function and survival. Significant progress has been made in recent years
Received in revised form 13 August 2009
towards a better understanding of the cellular and molecular basis of glucolipotoxicity in the beta cell. The
Accepted 13 August 2009
permissive effect of elevated glucose on the detrimental actions of fatty acids stems from the influence of
Available online 26 August 2009
glucose on intracellular fatty acid metabolism, promoting the synthesis of cellular lipids. The combination ofexcessive levels of fatty acids and glucose therefore leads to decreased insulin secretion, impaired insulin
Keywords:Fatty acid
gene expression, and beta-cell death by apoptosis, all of which probably have distinct underlying
mechanisms. Recent studies from our laboratory have identified several pathways implicated in fatty acid
Islet of Langerhans
inhibition of insulin gene expression, including the extracellular-regulated kinase (ERK1/2) pathway, the
metabolic sensor Per-Arnt-Sim kinase (PASK), and the ATF6 branch of the unfolded protein response. We
have also confirmed in vivo in rats that the decrease in insulin gene expression is an early defect whichprecedes any detectable abnormality in insulin secretion. While the role of glucolipotoxicity in humans isstill debated, the inhibitory effects of chronically elevated fatty acid levels has been clearly demonstrated inseveral studies, at least in individuals genetically predisposed to developing type 2 diabetes. It is thereforelikely that glucolipotoxicity contributes to beta-cell failure in type 2 diabetes as well as to the decline in beta-cell function observed after the onset of the disease.
2009 Elsevier B.V. All rights reserved.
aggravates metabolic perturbations, and so on. While elevated levelsof glucose or fatty acids can, by themselves, be demonstrated to have
Over the last 20 years, the central role of pancreatic beta-cell
detrimental effects on beta-cell function in many experimental
dysfunction in the development of type 2 diabetes has become
systems, the combination of both nutrients is synergistically harmful,
increasingly appreciated [1]. It is now generally accepted that when
which has led to the concept of glucolipotoxicity [7,8]. However,
insulin resistance develops in response to environmental cues such as
despite years of investigation and significant progress made in the
obesity, a subset of genetically predisposed individuals fails to
discovery of the underlying molecular and cellular mechanisms of
adequately compensate for the increased insulin demand, and beta-
glucolipotoxicity, its contribution to beta-cell failure in type 2 diabetes
cell failure ensues [2]. In addition, longitudinal studies in humans
remains debated. We speculate that this uncertainty stems from
have clearly demonstrated that beta-cell function deteriorates during
several reasons. First, by nature of their long-term design, experi-
the years following diagnosis of type 2 diabetes, regardless of the
ments to test cause-and-effect relationships between chronic meta-
therapeutic regimen [3,4]. Although the cause of this metabolic
bolic perturbations and functional outcomes are plagued with
deterioration is unknown, several hypotheses have been proposed.
confounding variables and therefore difficult to interpret. Second,
Amongst them, chronic hyperglycemia (glucotoxicity [5]), chronic
the inherent limitations of in vivo models have prompted the
dislipidemia (lipotoxicity [6]), or the combination of both (glucoli-
development of many in vitro systems to test the hypothesis and
potoxicity [7]), have been postulated to contribute to the worsening of
define its underlying mechanisms. As further discussed in this review,
beta-cell function over time, creating a vicious cycle by which
these systems also have important caveats. Third and perhaps most
metabolic abnormalities impair insulin secretion, which further
importantly, there is no clear consensus on the definition of the termglucolipotoxicity. While its root (toxicity) implies the presence of celldeath, it is often employed more loosely to refer to the functionaleffects of the combination of high glucose and elevated lipids on the
⁎ Corresponding author. CRCHUM, Technopole Angus, 2901 Rachel Est, Montreal, QC,
beta cell, for instance on insulin secretion or gene expression. Also,
H1W 4A4, Canada. Tel.: +1 514 890 8000x23603; fax: +1 514 412 7648.
E-mail address: [email protected] (V. Poitout).
while the concept of glucolipotoxicity implicitly refers to a chronic
1388-1981/$ – see front matter 2009 Elsevier B.V. All rights reserved.
doi:10.1016/j.bbalip.2009.08.006
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
situation, the notion of chronicity is variable, spanning from a few
transported into the mitochondria via the enzyme carnitine-
hours of ex vivo cell culture to many years in diabetic patients. This is
palmitoyl transferase-1 (CPT-1) for beta-oxidation, which has
particularly problematic since fatty acids have a dual and time-
essentially no functional consequences. In contrast, when both
dependent effect on beta-cell function, acutely stimulatory but
glucose and fatty acid concentrations are elevated, intracellular
chronically inhibitory. Thus, there are virtually as many definitions
metabolism of glucose leads to the formation of cataplerotic signals,
of the term glucolipotoxicity as groups studying it, which has created
such as citrate, and the generation of malonyl-CoA in the cytosol.
confusion in the field. For the purpose of this article, we propose to
Since fatty acid synthase activity is lower than that of acetyl-CoA
define glucolipotoxicity as the combined, deleterious effects of
carboxylase in the beta cell [33], the predominant effect of malonyl-
elevated glucose and fatty acid levels on pancreatic beta-cell function
CoA is to inhibit CPT-1 activity, which in turn blocks fatty acid
and/or survival. This review focuses on recent developments in the
oxidation and leads to accumulation of long-chain acyl-CoA esters
field of glucolipotoxicity from both in vitro and in vivo studies.
(LC-CoA) in the cytosol [7]. Accumulation of cytosolic LC-CoA, eitherdirectly or via generation of lipid-derived signals, adversely affects
2. Cellular and molecular mechanisms of glucolipotoxicity in the
beta-cell function [8]. In addition to its metabolic effects directing
fatty acid partitioning into esterification, glucose coordinatelyactivates the expression of genes involved in lipogenesis [34]. A
Considering the complexity of designing mechanistic studies in
key player in this mechanism is the enzyme AMP-activated protein
vivo to investigate the chronic effects of fuel oversupply, a number
kinase (AMPK), acting as a metabolic sensor that directs the beta cell
of in vitro models, using insulin-secreting cells and isolated islets,
into a "storage mode" in the face of nutrient oversupply [35], as it
have been employed to identify the cellular and molecular basis of
does in myocytes and hepatocytes [36]. Indeed, AMPK activity is
glucolipotoxicity. In these systems, prolonged exposure to elevated
inversely correlated with the glucose concentration [37] and is
levels of fatty acids is associated with inhibition of glucose-induced
stimulated by palmitate [38] in beta cells. Downstream of AMPK, the
insulin secretion [9–12], impairment of insulin gene expression
[13–18], and induction of cell death by apoptosis [19–28].
(SREBP1c), which regulates the expression of genes controlling
Importantly, several of these studies have provided evidence that
fatty acid synthesis [39], translates the metabolic signal sensed by
lipotoxicity only occurs in the presence of concomitantly elevated
AMPK into changes in gene expression, leading to enhanced
glucose levels [15,16,28], an observation also confirmed in vivo
lipogenesis. Glucose also increases the expression of liver X receptor
[29,30]. The biochemical basis for this permissive effect of glucose
(LXR) which then contributes to enhancing SREBP1c expression and
will be discussed first in this section, followed by a review of the
lipid synthesis [40].
mechanisms underlying the functional manifestations of glucolipo-
While it is now generally accepted that fatty acid partitioning
toxicity on the beta cell (insulin secretion, insulin gene expression,
towards esterification and cellular lipid synthesis underpins the
and cell survival).
cellular mechanisms of glucolipotoxicity in pancreatic beta cells, thenature of the lipid-derived metabolites directly responsible for the
2.1. Biochemical pathways and lipid intermediates implicated in
deleterious effects of fatty acids is still elusive. It is unlikely that
triglyceride accumulation itself might be the culprit, since triglycer-ides represent a relatively innocuous form of fat storage that can
The permissive effect of glucose on the deleterious actions of
actually protect against lipotoxicity [41]. Studies have shown that
chronic fatty acids stems from its influence on intracellular
monounsaturated fatty acids are less toxic and can actually protect
metabolism of fatty acids [31,32]. Prentki and Corkey [7] first
from the detrimental effects of unsaturated fatty acids because they
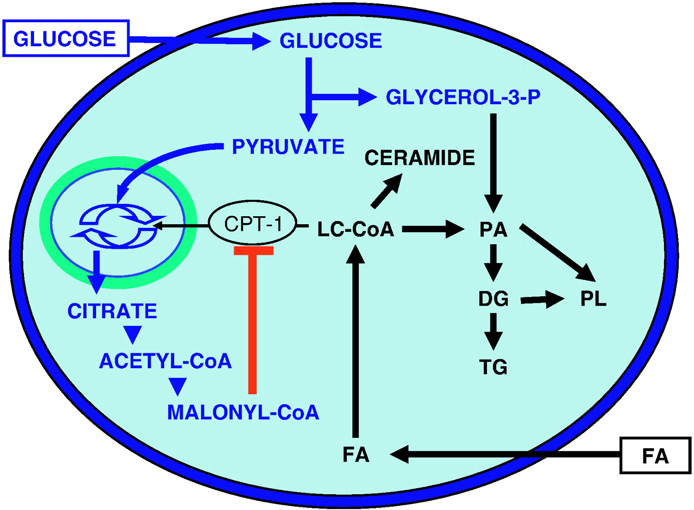
proposed that glucose determines fatty acid partitioning in pancre-
are more readily esterified into triglycerides [26,41]. Consistent with
atic beta cells (Fig. 1). At low glucose concentrations, fatty acids are
this notion is the observation that stearoyl CoA desaturase-1 (SCD1)protects from lipoapoptotic cell death induced by palmitate [42]. Infact, whereas deletion of SCD1 in mice improves insulin sensitivity[43], when introduced on the obese, leptin-deficient ob/ob back-ground the SCD1 deletion leads to a worsening of diabetes associatedwith triglyceride and cholesterol overload in islets [44].
Nolan and Prentki [45] and Prentki and Madiraju [46] have
proposed the elegant concept that increased glycerolipid/fatty acidcycling represents a mean by which the beta cell attempts to protectitself from nutrient oversupply while remaining fuel-responsive so asto be capable of releasing insulin in the face of increased demand. Inturn, the unintended consequence of this fuel detoxification mech-anism is the generation of harmful intermediates from increased fluxthrough the cycle. The question remains that if triglyceride accumu-lation is merely a marker of enhanced esterification flux but does notcause glucolipotoxicity by itself, then what are the lipid-derivedmolecules directly responsible for the impairment of beta-cellfunction? The role of intermediates of the esterification pathway(e.g. lysophosphatidic acid, phosphatidic acid, diacylglycerols) hasbeen suggested [2] but, to our knowledge, not formally demonstrated.
De novo synthesis of ceramide has been shown to play a role both in
Fig. 1. Effects of glucose on lipid partitioning in the beta cell. In the presence ofsimultaneously elevated levels of glucose and fatty acid (FA), the increase in cytosolic
fatty acid-induced beta-cell death [47] and fatty acid inhibition of
malonyl-CoA resulting from glucose metabolism inhibits the enzyme carnitine-
insulin gene expression [17] but not in the impairment of insulin
palmitoyl transferase-1 (CPT-1). Transport of long-chain acyl-CoA (LC-CoA) in the
secretion [48]. These observations illustrate an important point, which
mitochondria is reduced, and the esterification pathway is preferentially activated,
may in part explain why the lipid-derived intermediates mediating
leading to cytosolic accumulation of lipid-derived signaling molecules such as
glucolipotoxicity have remained elusive: the mechanisms underlying
ceramide, diglycerides (DG), phosphatidic acid (PA), phospholipids (PL), andtriglycerides (TG).
the various functional manifestations of glucolipotoxicity are likely
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
distinct. For example, accumulation of ceramide impairs insulin gene
insulin secretion [65,66] and that UCP2 KO animals on a mixed genetic
expression and, under certain circumstances, induces cell death,
background have increased circulating insulin levels and are protected
without affecting insulin secretion. Therefore, our view is that the full
from diabetes [63,67]. This contention has been recently challenged by
array of functional defects associated with glucolipotoxic conditions is
the observation that KO of UCP2 on 3 different congenic backgrounds in
due to the generation of several intracellular metabolites acting on
the mouse leads to oxidative stress and impaired insulin secretion [68].
various signaling pathways and cellular functions rather than to a
Thus, the increase in UCP2 expression observed in islets after high-fat
feeding in rodents [30,66] or exposure to fatty acids in vitro [69,70]
While most studies investigating the mechanisms of glucolipo-
likely represents a cellular defense mechanism against fuel overload and
toxicity in the beta cell have focused on the esterification pathway and
oxidative stress rather than a deleterious response. Consistent with this
triglyceride synthesis, cholesterol metabolism has recently been
possibility is the observation that transgenic overexpression of UCP2
shown to also play an important role. Exposure of beta cells to
does not alter mitochondrial function or glucose-induced insulin
oxidized low-density lipoproteins (LDL) induces apoptosis [49] and
secretion but decreases reactive oxygen species production [71].
decreases insulin gene expression [50], whereas native LDL particles
Overall, it appears unlikely that an increase in UCP2 expression in
have no effect and high-density lipoproteins (HDL) are protective.
response to fatty acids represents a causal mechanism of the
Beta-cell specific knock-out (KO) of the ATP-binding cassette
impairment of insulin secretion under glucolipotoxic conditions.
transporter subfamily A member 1 (ABCA1), which mediates reverse
Activation of the lipid-regulated isoform PKCɛ has also been
cholesterol efflux, results in increased cellular cholesterol content and
suggested as a possible candidate signaling molecule underlying the
impaired insulin secretion downstream of glucose metabolism,
decrease in insulin secretion in glucolipotoxicity. Work by Schmitz-
probably at the level of insulin exocytosis [51]. In addition, the ability
Peiffer et al. [72] has shown that the normalization of glucose tolerance
of the thiazolidinedione rosiglitazone to improve glucose tolerance in
in PKCɛ KO mice under high-fat feeding was due to improved insulin
high-fat diet fed mice requires a functional ABCA1 in beta cells [51].
secretion. Further, they demonstrated that islets isolated from PKCɛ
Finally, forcing cholesterol synthesis in beta cells by transgenic
knock-out mice were protected from the deleterious effects of fatty acids
overexpression of SREBP2 under the rat insulin promoter results in
on insulin secretion in vitro and that inhibition of PKCɛ was capable of
a severe loss of beta-cell mass and a diabetic phenotype [52]. Since
restoring insulin secretion in islets from db/db mice [72]. More recently,
LXR regulates ABCA1 expression [51] and is itself directly regulated by
this group has shown that the improvement in insulin secretion in PKCɛ
glucose [53], glucose therefore coordinately increases fatty acid
knock-out islets in the face of glucolipotoxicity was due to selective
esterification and intracellular cholesterol synthesis.
restoration of the amplifying pathway of insulin release, probably due to
The premise to the hypotheses described above that intermediates
the generation of a lipolytic intermediate [73]. Interestingly, this is
generated during triglyceride or cholesterol synthesis are mechanisti-
consistent with the concept proposed by Peyot et al. [74] that lipolysis-
cally involved in glucolipotoxicity is that extracellular fatty acids are
generated signals contribute to the regulation of insulin secretion and
first transported across the plasma membrane and act intracellularly.
that, more generally, glycerolipid/fatty acid cycling in the beta cell
This concept has been challenged by the deorphanization of the G-
provides essential coupling factors for insulin secretion but becomes
protein coupled receptor GPR40 [54,55]. GPR40 is specifically expressed
detrimental under conditions of fuel oversupply [45,46].
in pancreatic beta cells and is activated by long-chain fatty acids, which
Finally, evidence suggests that fatty acids might alter one or more
raises the possibility that some of the functional effects of fatty acids on
late steps of insulin exocytosis in beta cells. Kato et al. [75] have shown
the beta-cell might be mediated by activation of a cell surface receptor.
that expression of granuphilin, an effector of the small GTP-binding
Consistent with this possibility, a role for GPR40 in mediating fatty acid
protein Rab27a, which plays a key role in the docking of insulin
inhibition of insulin secretion has been suggested by the observation
secretory granules to the plasma membrane, is increased in islets
that islets from GPR40 KO mice are insensitive to the inhibitory effects
exposed to palmitate as a consequence of upregulation of SREBP1c. This
of prolonged fatty acids [56]. Using a different line of GPR40 KO mice,
in turn inhibits insulin secretion in response to fuel and non-fuel stimuli.
we were unable to reproduce these findings and found that deletion of
In addition, Olofsson et al. [76] demonstrated that prolonged exposure
the receptor does not protect islets from fatty acid inhibition of glucose-
of mouse islets to glucose and fatty acids inhibited insulin secretion at a
induced insulin secretion [57]. In addition, subsequent studies also
very late stage of exocytosis by interfering with the release of insulin at
using whole-body KO found that GPR40 deletion did not protect mice
the fusion pore. These findings suggest that the mechanisms by which
from high-fat diet-induced glucose intolerance [58,59]. This conclusion
fatty acids affect insulin secretion might, at least in part, lie at the level of
was further supported by the observation that small molecule GPR40
the exocytotic machinery and, consequently, impair insulin secretion in
agonists improved glucose tolerance in mice with high-fat diet-induced
response not only to glucose but also to other secretagogues.
obesity [60]. Therefore, we do not favor the view that GPR40 plays amajor role in the mechanisms of glucolipotoxicity in the beta cell.
2.2.2. Fatty acid impairment of insulin gene expression
We [15–18,77] and others [13,14] have shown that prolonged
2.2. Mechanisms underlying the functional manifestations of
exposure to fatty acids impairs insulin gene expression in the
presence of high glucose. The mechanisms whereby fatty acids affectinsulin gene expression are distinct from those by which they impair
2.2.1. Fatty acid impairment of insulin secretion
insulin secretion. First, whereas both palmitate and oleate inhibit
Prolonged exposure of beta cells to fatty acids in vitro inhibits
insulin secretion, only palmitate affects insulin gene expression [48].
glucose-stimulated insulin secretion [9–12], a phenomenon also
This is due to the fact that only palmitate can serve as a substrate for
observed in vivo in rats [61] and humans [62]. In recent years, several
de novo ceramide synthesis [17]. The transcriptional mechanisms by
potential mechanisms have been investigated, including upregulation
which palmitate inhibits insulin gene expression do not involve
of uncoupling protein 2 (UCP2), activation of the novel isoform of
changes in insulin mRNA stability but, rather, inhibition of glucose-
protein kinase C PKCɛ, and late exocytotic events.
induced insulin promoter activity [17]. This is associated with
UCP2 is a ubiquitously expressed mitochondrial carrier which has
decreased binding activity of the transcription factors pancreas–
been suggested to uncouple the respiratory chain from ATP synthesis
duodenum homeobox 1 (PDX-1) and MafA [18]. PDX-1 is affected in
[63], although its biological functions are still unclear [64]. Initial
its ability to translocate to the nucleus, whereas MafA is affected at
evidence suggested that UCP2 might modulate insulin secretion and
the level of its expression [18]. This is in contrast to the mechanisms
thereby play a role in glucolipotoxicity. This was based on the
of glucotoxicity, which involve post-translational modifications of
observations that increasing UCP2 expression in beta cells impairs
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
The mechanisms whereby ceramide generation from palmitate
contribute to the overall decrease in insulin gene expression [84]
impairs PDX-1 subcellular localization and MafA expression are
(Fig. 2). Although our initial study revealed that palmitate mostly
unknown, although recent studies have identified potential candi-
affects PDX-1 in its subcellular localization rather than its whole-cell
dates. The c-jun NH2-terminal kinase JNK is a known target of
expression levels [18], overexpression of a kinase dead mutant of
ceramide [79] and can repress insulin gene transcription both via c-
PASK also reduces PDX-1 mRNA levels. This suggests that reduction
jun-dependent inhibition of E1-mediated transcription [80,81] and
of PDX-1 expression might also contribute to decreasing its binding
c-jun independent inhibition of PDX-1 binding [82]. In addition,
activity under glucolipotoxic conditions. Whether PASK can directly
Solinas et al. [83] have shown that palmitate activates JNK in beta
phosphorylate PDX-1 and, thereby, alter its nuclear translocation is
cells and that the resulting phosphorylation of insulin receptor
unknown and currently under investigation. Recently, expression of
substrates 1 and 2 at sites that impair insulin signaling decreases
the CAAT enhancer-binding protein β (C/EBPβ), a negative regulator
insulin gene transcription.
of insulin gene transcription [89], has been shown to increase in beta
Recent studies in our laboratory have also attempted to identify
cells in response to fatty acids [90]. Interestingly, we also observed a
the signaling mechanisms implicated in palmitate inhibition of
marked increase in C/EBPβ mRNA levels upon overexpression of the
insulin gene expression. First, we have shown that palmitate
dominant-negative PASK mutant in MIN6 cells [84]. This raises the
enhances glucose-induced phosphorylation of the extracellular-
possibility that, as demonstrated under glucotoxic conditions [91],
regulated kinases (ERK) 1/2 and that pharmacological inhibition of
C/EBPβ binds to the transcription factor nuclear factor of activated T
ERK1/2 partially restores insulin gene expression in insulin-
cells (NFAT) on the insulin promoter and thereby inhibits MafA
secreting cells and isolated islets exposed to palmitate or ceramide
binding activity.
[84]. Second, we have observed that palmitate blocks the induction
A role for the unfolded protein response (UPR) and endoplasmic
of the Per-Arnt-Sim kinase (PASK) by glucose [84]. PASK is an
reticulum (ER) stress in beta-cell failure has received considerable
evolutionarily conserved serine/threonine protein kinase, containing
attention in the past few years, in part because the beta cell's intense
a PAS domain sensitive to the intracellular environment which
secretory activity makes it particularly susceptible to perturbations of
regulates the kinase domain to transduce the signal [85]. In budding
ER homeostasis [92]. As discussed in more details in the next section,
yeast, it coordinates sugar storage and protein synthesis with
markers of ER stress have been shown to be induced by prolonged
carbohydrate availability [86]. In mammals, it has been demonstrat-
exposure to fatty acids in several studies [93–101]. In most cases, the
ed to be an important regulator of glycogen synthase and cellular
strong induction of ER stress markers in response to fatty acids is
energy balance [87]. In pancreatic beta cells, PASK is required for
associated with apoptosis. Under our culture conditions of isolated rat
glucose-induced insulin gene transcription [88]. In our recent study
islets in the presence of glucose and palmitate, which are not
[84], we observed that overexpression of PASK prevents the
associated with significant cell death [84,102], we have not been
inhibitory effect of palmitate on insulin mRNA and PDX-1 mRNA
able to detect any activation of the inositol requiring ER-to-nucleus
and protein expression in MIN6 cells. In addition, adenoviral-
signal kinase (IRE) or protein kinase R-like ER kinase (PERK) branches
mediated overexpression of wild-type PASK increased, whereas a
of the UPR (unpublished data). In contrast, we have observed cleavage
kinase dead mutant of PASK acting as a dominant negative
of the transcription factor ATF6 under these conditions. Since ATF6 is a
decreased, insulin mRNA and PDX-1 protein expression in islets.
negative regulator of insulin gene transcription [103], these prelim-
Interestingly, the PASK pathway appears to be independent from the
inary results led us to hypothesize that an early activation of the ATF6
ERK1/2 pathway and to have no effect on MafA expression in our
branch of the unfolded protein response upon exposure to fatty acids
system, suggesting that at least 3 independent signaling arms
might represent a protective mechanism whereby the beta cellattempts to further decrease the load to the ER by inhibiting insulingene expression. This would occur as part of the unfolded proteinresponse, before overt ER stress and associated apoptosis develops. Inlater stages of more severe ER stress associated with cell death, it ispossible that alterations in PDX-1 function [96,104] or insulin mRNAstability [105] also contribute to the decrease in insulin geneexpression.
Overall, available data regarding the mechanisms of fatty acid
inhibition of the insulin gene reveal a complex picture which appearsto involve several independent pathways that all concur to decreaseits expression, which is an early, and possibly protective, response ofthe beta cell in the face of nutrient oversupply (Fig. 2). Importantly,the decrease in insulin gene expression under glucolipotoxic condi-tions is also observed in vivo ([77]; see Section 3).
2.2.3. Fatty acid induction of beta-cell death
Saturated fatty acids can induce beta-cell death by apoptosis in
the presence of high glucose [22,26,28], whereas unsaturated fattyacids are usually protective [21,22,28]. As mentioned above, thisdifference is likely due to the greater ability of unsaturated fattyacids to form intracellular triglycerides [21,41,42]. Several mechan-
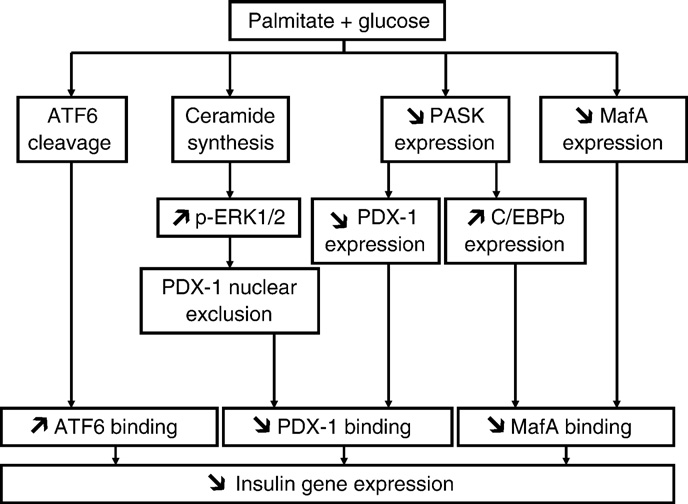
Fig. 2. Working model of the mechanisms of fatty acid inhibition of insulin gene
isms have been implicated, including ceramide formation
expression. Several signaling pathways are activated in beta cells in the presence of
[20,23,26,47], oxidative stress [25,27,106,107], and inflammation
simultaneously elevated levels of palmitate and glucose. First, de novo ceramidesynthesis [17] leads to sustained activation of ERK1/2 [82] and exclusion of PDX-1 from
[108]. Recently, as mentioned above, considerable evidence has been
the nuclear compartment [18]. Second, palmitate blocks glucose-induction of PASK
provided in support of a role for the UPR and ER stress in saturated
expression, which results in decreased PDX-1 expression and increased C/EBPβ
fatty acid-induced cell death ([93–101] and reviewed in [59]). The
expression [82]. Third, palmitate decreases MafA expression [18]. These 3 pathways
mechanisms by which saturated fatty acids such as palmitate induce
result in decreased binding activities of PDX-1 and MafA on the insulin promoter. In
ER stress are thought to involve depletion of ER calcium stores
addition, palmitate induces the cleavage of ATF6, which also represses insulin genetranscription (our unpublished data).
[99,101] and result in the activation of JNK [99,100], although JNK
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
activation can, under some conditions, be detected prior to the
3. In vivo studies
appearance of ER stress [98]. Interestingly, palmitate was shown toinduce a rapid degradation of carboxypeptidase E, which resulted
3.1. Rodent models of glucolipotoxicity
not only in altered proinsulin maturation, but also in ER stress andapoptosis [109]. The changes in CPE levels were demonstrated to
For the reasons described above, the findings of in vitro studies
occur prior to the development of any sign of ER stress and to
should be confirmed in vivo before they can be extrapolated to
require palmitate metabolism and calcium influx, although the
physiological or pathological situations. In this regard, pioneering
precise mechanisms by which palmitate initiates CPE degradation
studies by the group of Unger in the Zucker diabetic fatty (ZDF) rat were
remain to be clarified [109]. Of note, however, a study by Lai et al.
instrumental in establishing the concept of lipotoxicity and identifying
[110] using insulin-secreting cells and isolated islets provided
some of its basic mechanisms (reviewed in [120]). In particular, these
evidence that palmitate-induced apoptosis can also occur in the
studies first identified the key role for ceramide as an intracellular
absence of detectable ER stress. Finally, markers of ER stress are
mediator of glucolipotoxicity. Thus, in this model, accumulation of
increased in pancreatic sections of type 2 diabetic patients [111].
intra-islet ceramide is detected prior to beta-cell dysfunction [121] and
These observations raise the question as to whether fatty acid-
inhibition of ceramide synthesis prevents beta-cell death [47]. In more
induced apoptosis in beta cells is primarily mediated by ER stress or
recent studies, the beneficial effects of pharmacological inhibition of
the mitochondrial death pathway. Intrinsic defects in mitochondrial
sphingolipid synthesis on beta-cell function and diabetes progression
function have been well documented under conditions of nutrient
have been confirmed not only in the ZDF rat but also in other rodent
overload [112], and perturbations in mitochondrial permeability are
models [122–124]. However, since ceramide is also implicated in the
observed early in the development of fatty acid-induced cell death in
mechanisms of insulin resistance [123], it is difficult in these in vivo
beta cells [113]. Luciani et al. [114] have recently shown that depletion
studies to distinguish between the effects of the treatment on insulin
of ER calcium stores under conditions of ER stress can lead secondarily
sensitivity and those on beta-cell function.
to mitochondrial dysfunction, suggesting that perhaps under gluco-
Non-genetic models of glucolipotoxicity have been developed
lipotoxic conditions ER stress is a primary event which leads to
and most often use prolonged infusions of Intralipid, a soybean oil
triggering of several proapoptotic pathways, including mitochondrial-
emulsion which generates a mixture of mostly unsaturated fatty
mediated cell death.
acids [125] when co-injected with heparin. In these models, the
Finally, a recent study by Lovis et al. [115] has shown that
effects of Intralipid or fatty acid infusion on beta-cell function have
increased expression of the microRNAs miR34a and miR146 also
been inconsistent, leading to either unaffected [77], enhanced
contributes directly to palmitate-induced cell death in insulin-
[126,127], or reduced [9,61,128,129] insulin secretion. These dis-
secreting cells and isolated islets, and the overall role of microRNAs
crepancies are likely due to differences in strain, sex, age, or infusion
in glucolipotoxicity will hopefully become clearer as progress towards
rates. For instance, Mason et al. [61] and Goh et al. [128] suggested
understanding their implications in beta-cell function continues to be
that female Wistar rats are more susceptible to the deleterious
effects of prolonged high fatty acid levels, and Steil et al. [127] haveobserved that a 96-h Intralipid infusion did not affect insulin
2.3. Limitations of in vitro studies of glucolipotoxicity
secretion in male Sprague-Dawley rats. The influence of geneticpredisposition on the insulin secretory response to excessive fatty
While in vitro models using insulin-secreting cells and isolated
acid levels is also illustrated by the observation that insulin secretion
islets have proven extremely valuable in dissecting the cellular and
is impaired to a greater extent in heterozygous lean ZDF rats than in
molecular mechanisms of glucolipotoxicity, they also have significant
Wistar rats after Intralipid infusion [128]. Recent studies in our
limitations which should be borne in mind when interpreting the
laboratory also highlight the importance of the age of the animals in
results obtained in these systems. First, there appears to be species-
the response to chronic fuel overload. In a first study, we infused 8-
related differences in the sensitivity to fatty acid-induced cell death
week-old male Wistar rats alternatively with glucose for 4 h and
[110]. For instance, whereas a 24-h exposure of human islets to
Intralipid + heparin for 4 h, for a total of 72 h [77]. Hyperglycemic
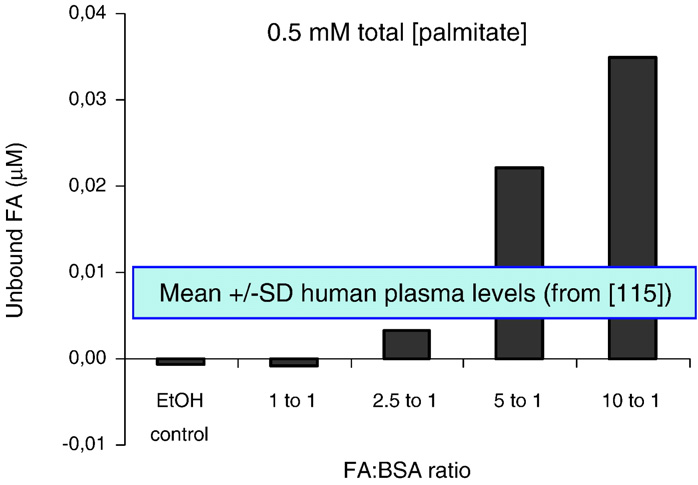
elevated glucose and palmitate is sufficient to observe apoptosis [28],we have not detected any cell death in rat islets after 72 h of cultureunder similar conditions [17,48,84]. Second, the concentrations offatty acids used in vitro vary amongst publications. The keydeterminant of fatty acid potency is the fraction that is unbound toBovine Serum Albumin (BSA) which depends on the molar ratio offatty acids to albumin as well as the mode of preparation. Using afluorescent probe that specifically measures the unbound fraction offatty acids [116], we observed that when palmitate at a totalconcentration of 0.5 mM was pre-complexed to BSA with a fattyacid-to-albumin molar ratio of 5:1, the unbound concentration is in therange of 200 nM (Fig. 3), which represents approximately 3 times theunbound concentration measured in the plasma of lean individuals bythe same method [117]. Finally, the concentrations of fatty acids in thevicinity of the beta cells in vivo are unknown and are probablydetermined by several different factors, including the activity oflipoprotein lipase, which accounts for some of the local delivery offatty acids to the cells [118]. In fact, it is likely that lipoprotein lipaseactivity is an important control point for fatty acid delivery to betacells, since both beta-cell specific deletion and overexpression of itsgene in the mouse impair glucose homeostasis and insulin secretion
Fig. 3. Concentrations of unbound fatty acids (FA) in solution as a function of the fattyacid-to-BSA ratio for a fixed total palmitate concentration of 0.5 mM. Unbound fatty
[119]. Thus, the results of in vitro experiments using fatty acids should
acids were measured using the fluorescent probe ADIFAB [114]. Data are the average of
be interpreted with caution, particularly when marked cytotoxicity is
2 independent experiments. Also represented are the mean ± SD of unbound FA levels
measured in human plasma using the same method, from Lovis et al. [115].
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
clamps performed at the end of the infusion failed to detect any
by Intralipid infusion in vivo in humans, suggesting the possible
effects of the glucose + Intralipid infusion regimen on insulin
contribution of oxidative stress [139].
secretion in vivo, as compared to control, saline-infused animals.
Finally, the group of Cusi and De Fronzo has carried out a series of
Similarly, insulin secretion in response to glucose in isolated islets
studies in non-diabetic subjects with and without family history of
was unaffected. In animals infused with glucose only, we observed
type 2 diabetes which clearly highlights the importance of genetic
an increase in insulin mRNA levels, PDX-1 nuclear localization, and
predisposition on the effects of chronically elevated fatty acids in
PDX-1 binding to the endogenous insulin gene promoter in islets. In
humans. They showed that a 4-day Intralipid infusion enhances
contrast, in islets from animals infused with glucose + Intralipid,
insulin secretion (taking into account insulin sensitivity) in control
insulin mRNA levels were reduced, PDX-1 localization was shifted
subjects but inhibits glucose-induced insulin secretion in individuals
towards the cytosol, and occupancy of the endogenous insulin
with a family history of type 2 diabetes [140]. This suggests that the
promoter by PDX-1 was markedly diminished [77]. These results
genetic predisposition to developing type 2 diabetes might be
demonstrate that fatty acid inhibition of the insulin gene also occurs
dependent, at least in part, on the ability of the beta cell to increase
in vivo and represents an early defect that can be detected prior to
insulin secretion in response to elevated fatty acid levels. Importantly,
any alteration in insulin secretion. The lack of effect of the infusion
treatment of susceptible subjects with Acipimox to decrease circulat-
on insulin secretion in 8-week-old rats prompted us to assess
ing fatty acid levels ameliorates insulin secretion [141].
whether older animals would be more susceptible to nutrientoverload. To test this possibility, we recently conducted a second
study in which glucose and Intralipid were infused simultaneouslyand continuously for 72 h to either 8-week-old or 6-month-old
In recent years, major progress has been made towards a better
Wistar rats (unpublished results). As in our first study, this infusion
understanding of the cellular and molecular mechanisms of
regimen did not alter insulin secretion in 8-week-old rats, as
glucolipotoxicity in the beta cell. The biochemical basis for the
assessed by hyperglycemic clamps at the end of the infusion. In
permissive effect of elevated glucose on the deleterious actions of
marked contrast, infusion of glucose + Intralipid in 6-month-old rats
fatty acids is better delineated; the mechanisms by which the
resulted in marked insulin resistance which was not adequately
combination of excessive levels of fatty acids and glucose alter beta
compensated for by a sufficient increase in insulin secretion in vivo
cell function are beginning to be unraveled; and it is becoming clear
and in defective insulin secretion in vitro in isolated islets. The
that the various functional effects of fatty acids (i.e., decreased
results from these two studies yield two important conclusions. First,
insulin secretion, impaired insulin gene expression, and beta-cell
defective insulin gene expression under glucolipotoxic conditions
death by apoptosis) have different underlying mechanisms. Despite
occurs in vivo and precedes abnormalities in insulin secretion. This
significant progress, however, a number of important questions
confirms the physiological relevance of our previous in vitro findings
remain. While it is now clear that triglyceride accumulation is more
[17,18] and suggests that impaired insulin gene transcription might
a symptom than a cause of glucolipotoxicity, the nature of the lipid-
represent an early defect in nutrient-induced beta-cell failure.
derived intermediates directly responsible for the detrimental
Second, young rats are resistant to the effects of nutrient oversupply,
effects of fatty acids is still elusive. In that regard, a role for
and such studies are probably better conducted in older animals,
cholesterol accumulation is also likely. Amongst the several
which more closely resemble the typical setting of type 2 diabetes in
candidates recently proposed to explain fatty acid inhibition of
humans. Whether or not this age-dependent susceptibility to
insulin secretion, the role of UCP2 has become unclear, while
nutrient oversupply is related to the reduced beta-cell proliferative
convincing evidence seems to implicate the novel isoform PKCɛ as
capacity in older rodents [130,131] is unknown and currently under
well as late exocytotic events. Regarding fatty acid impairment of
the insulin gene, a complex picture has emerged which includesprolonged activation of ERK1/2 via de novo ceramide synthesis,downregulation of PASK, and altered binding activities of the
3.2. Studies in humans
transcription factors PDX-1, MafA, and C/EBPβ. The role of theUPR under conditions of mild glucolipotoxicity (i.e., not associated
As in experimental animals, studies examining the effects of
with cell death) appears limited, although our current hypothesis is
prolonged fatty acids on insulin secretion in humans have led to
that early activation of ATF6 represses insulin gene transcription
conflicting results. Initial reports from Boden et al. indicated that a 48-
and thereby contributes to the reduction in proinsulin biosynthesis
h lipid infusion induces an appropriate insulin secretory response in
in an attempt to decrease the load to the ER. As conditions
healthy subjects [132] but is defective in type 2 diabetic patients
deteriorate, unresolved and sustained unfolded protein response
[133]. In contrast, Carpentier et al. [134] showed in non-diabetic
likely leads to ER stress and, consequently, to beta-cell apoptosis
individuals that an acute (90-min) lipid infusion elicits an increase in
under severe glucolipotoxic conditions. The necessity to confirm in
insulin secretion which disappears when the infusion is prolonged for
vitro findings under physiological conditions has prompted several
48 h. The loss of insulin secretion is specific to the response to glucose,
groups, including ours, to address these questions in in vivo models.
as the response to arginine remains normal [135]. The same group
Our studies have confirmed that the decrease in insulin gene
further showed that obese, but not diabetic, subjects are susceptible to
expression is an early defect, which precedes any detectable
the inhibitory effect of lipids on glucose-induced insulin secretion
abnormality in insulin secretion, and have established that pro-
[136]. Importantly, the increase in insulin secretion observed in non-
longed infusions of glucose and Intralipid impairs beta-cell function
diabetic subjects in response to a 24-h glucose infusion does not occur
in old, but not young, animals, raising caution on the use of younger
if lipids are infused simultaneously with glucose [137]. Xiao et al.
rodents to examine mechanisms of beta-cell failure. While still
[138] confirmed that fatty acids also alter beta-cell function in obese
debated, the role of glucolipotoxicity in humans has been clearly
individuals when ingested orally, and observed interesting differences
demonstrated in several studies, at least in individuals genetically
between saturated and polyunsaturated fatty acids. While polyunsat-
predisposed to developing type 2 diabetes.
urated fatty acids impair insulin secretion directly, saturated fatty
We propose that the uncertainties regarding the role of
acids induce insulin resistance which was not adequately compen-
glucolipotoxicity and its manifestations stem from the fact that it is
sated for by an increase in beta-cell function [138]. The same group
being considered, as its name implies, as a deleterious phenomenon,
further observed that concomitant administration of the antioxidant
while in fact the beta cell's response to nutrient excess likely
taurine improved insulin resistance and beta-cell dysfunction induced
represents a continuum encompassing all stages of beta-cell
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
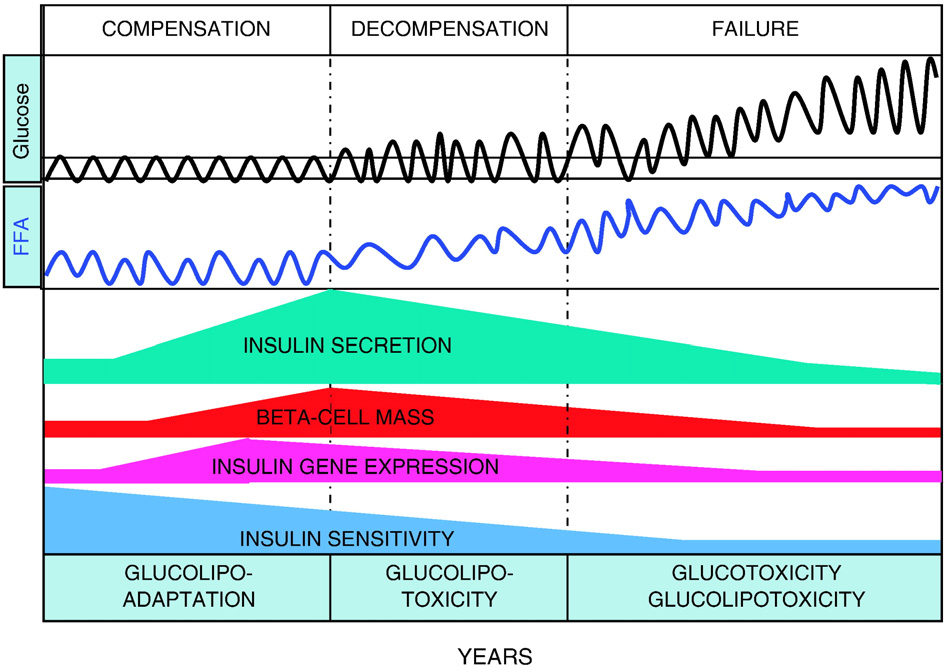
Fig. 4. Hypothetical representation of the progression from beta-cell compensation to failure in the face of obesity-induced insulin resistance and the role of glucolipotoxicity.
According to this hypothesis, the decrease in insulin sensitivity is initially matched by a marked increase in insulin secretion, insulin gene expression, and beta-cell mass. At thisstage, the beta cell adapts to nutrient oversupply by switching to preferential utilization of fatty acids, as part of the compensatory response (glucolipoadaptation [2]). In geneticallypredisposed individuals, the beta cell eventually becomes unable to further compensate and glucolipoadaptation evolves towards glucolipotoxicity, in which excursions of bloodglucose levels outside of the normal range become permissive for the detrimental effects of elevated fatty acids. This phase is characterized by an early loss of insulin gene expression,decreased insulin secretion (relative to the degree of insulin resistance), and reduced beta-cell mass. Finally, beta-cell failure occurs when glucose levels are permanently in thehyperglycemic range. At that stage, both glucotoxicity and glucolipotoxicity contribute to the continued deterioration of beta-cell function.
compensation and beta-cell failure. In that sense, some of the early
rodents, but additional investigation is necessary to ascertain the
manifestations of glucolipotoxicity should actually be considered as a
precise contribution of glucolipotoxicity to the pathogenesis of type 2
positive response and would be more appropriately named «gluco-
diabetes in humans.
lipoadaptation», as proposed by Prentki and Nolan [2]. Examples ofsuch adaptive responses are the early decrease in insulin gene
expression, as an attempt to protect the ER from overload [77], or theincrease in UCP2 expression, as a defense mechanism against
Work performed in our laboratory was supported by the US
oxidative stress [68].
National Institutes of Health (R01-DK58096 from NIDDK) and the
The hypothesis that glucolipotoxicity represents a continuum from
Canadian Institutes of Health Research (MOP 77686). V.P. holds the
an adaptive response to a deleterious outcome is illustrated in Fig. 4.
Canada Research Chair in Diabetes and Pancreatic Beta-cell Function.
According to this view, in normoglycemic individuals experiencing
G.F. is supported by a post-doctoral fellowship from the Canadian
weight gain, the beta cell mounts a compensatory response to counter
Diabetes Association. B.Z. is supported by the Montreal Diabetes
insulin resistance associated with obesity. This response involves
Research Center/Merck Frosst post-doctoral fellowship.
coordinated increases in beta-cell mass, insulin biosynthesis, andinsulin secretion and likely relies on an enhanced responsiveness tofatty acids [142,143]. The magnitude of the compensatory beta-cell
response is probably genetically determined and, in turn, is a major
[1] R.A. DeFronzo, From the triumvirate to the ominous octet: a new paradigm for
determinant of the long-term ability of an individual to maintain
the treatment of type 2 diabetes mellitus, Diabetes 58 (2009) 773–795.
glucose homeostasis in the face of insulin resistance. In contrast, in
[2] M. Prentki, C.J. Nolan, Islet beta cell failure in type 2 diabetes, J. Clin. Invest. 116
genetically predisposed individuals, beta-cell compensation eventu-
(2006) 1802–1812.
[3] U.K. prospective diabetes study 16, Overview of 6 years' therapy of type II
ally becomes insufficient and the beta cell is no longer able to sustain a
diabetes: a progressive disease. U.K. Prospective Diabetes Study Group, Diabetes
secretory response that matches the demand imposed by insulin
44 (1995) 1249–1258.
resistance. It is probably during this decompensation phase that
[4] S.E. Kahn, S.M. Haffner, M.A. Heise, W.H. Herman, R.R. Holman, N.P. Jones, B.G.
Kravitz, J.M. Lachin, M.C. O'Neill, B. Zinman, G. Viberti, the ADOPT Study
glucolipotoxicity plays a major role, in that hyperglycemia is the
Group, Glycemic durability of rosiglitazone, metformin, or glyburide mono-
permissive factor by which elevated fatty acids affect beta-cell
therapy, N. Engl. J. Med. 355 (2006) 2427–2443.
function. Our data suggest that one of the first functional defect at
[5] R.P. Robertson, J.S. Harmon, Y. Tanaka, G. Sacchi, P.O.T. Tran, C.E. Gleason, V.
this stage is a decrease in insulin gene expression, which likely
Poitout, Glucose toxicity of the beta-cell: cellular and molecular mechanisms, in:D. Le Roith, S.I. Taylor, J.M. Olefsky (Eds.), Diabetes Mellitus. A Fundamental and
contributes to eventual beta-cell failure since maintenance of
Clinical Text, 2nd Ed, Lippincott Williams & Wilkins, Philadelphia, 2000,
adequate intracellular stores of insulin is necessary to sustain
pp. 125–132.
increased secretory demand [144]. Beta-cell decompensation evolves
[6] R.H. Unger, Lipotoxicity in the pathogenesis of obesity-dependent NIDDM.
Genetic and clinical implications, Diabetes 44 (1995) 863–870.
towards beta-cell failure when fasting hyperglycemia occurs. At this
[7] M. Prentki, B.E. Corkey, Are the β-cell signaling molecules malonyl-CoA and
stage, it is likely that both glucotoxicity and glucolipotoxicity
cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and
contribute to the decline in insulin secretion observed over time
NIDDM? Diabetes 45 (1996) 273–283.
[8] V. Poitout, R.P. Robertson, Minireview: secondary beta-cell failure in type 2
during the years following diagnosis of type 2 diabetes [3]. This model
diabetes—a convergence of glucotoxicity and lipotoxicity, Endocrinology 143
is based on extensive experimental evidence obtained in vitro and in
(2002) 339–342.
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
[9] Y. Sako, V.E. Grill, A 48-hour lipid infusion in the rat time-dependently inhibits
[35] N. Ruderman, M. Prentki, AMP kinase and malonyl-CoA: targets for therapy of
glucose-induced insulin secretion and β-cell oxidation through a process likely
the metabolic syndrome, Nat. Rev., Drug Discov. 3 (2004) 340–351.
coupled to fatty acid oxidation, Endocrinology 127 (1990) 1580–1589.
[36] D.G. Hardie, Minireview: the AMP-activated protein kinase cascade: the key
[10] M.L. Elks, Chronic perfusion of rat islets with palmitate suppresses glucose-
sensor of cellular energy status, Endocrinology 144 (2003) 5179–5183.
stimulated insulin release, Endocrinology 133 (1993) 208–214.
[37] I.P. Salt, G. Johnson, S.J. Ashcroft, D.G. Hardie, AMP-activated protein kinase is
[11] Y.-P. Zhou, V. Grill, Long term exposure to fatty acids and ketones inhibits B-cell
activated by low glucose in cell lines derived from pancreatic beta-cells, and may
functions in human pancreatic islets of Langerhans, J. Clin. Endocrinol. Metab. 80
regulate insulin release, Biochem. J. 335 (1998) 533–539.
(1995) 1584–1590.
[38] X. Wang, L. Zhou, G. Li, T. Luo, Y. Gu, L. Qian, X. Fu, F. Li, J. Li, M. Luo, Palmitate
[12] Y.P. Zhou, V.E. Grill, Long-term exposure of rat pancreatic islets to fatty acids
activates AMP-activated protein kinase and regulates insulin secretion from beta
inhibits glucose-induced insulin secretion and biosynthesis through a glucose–
cells, Biochem. Biophys. Res. Commun. 352 (2007) 463–468.
fatty acid cycle, J. Clin. Invest. 93 (1994) 870–876.
[39] F. Foufelle, P. Ferre, New perspectives in the regulation of hepatic glycolytic and
[13] S. Gremlich, C. Bonny, G. Waeber, B. Thorens, Fatty acids decrease IDX-1
lipogenic genes by insulin and glucose: a role for the transcription factor sterol
expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and
regulatory element binding protein-1c, Biochem. J. 366 (2002) 377–391.
somatostatin levels, J. Biol. Chem. 272 (1997) 30261–30269.
[40] S.S. Choe, A.H. Choi, J.W. Lee, K.H. Kim, J.J. Chung, J. Park, K.M. Lee, K.G. Park, I.K.
[14] B. Ritz-Laser, P. Meda, I. Constant, N. Klages, A. Charollais, A. Morales, C. Magnan,
Lee, J.B. Kim, Chronic activation of liver X receptor induces beta-cell apoptosis
A. Ktorza, J. Philippe, Glucose-induced preproinsulin gene expression is inhibited
through hyperactivation of lipogenesis: liver X receptor-mediated lipotoxicity in
by the free-fatty acid palmitate, Endocrinology 140 (1999) 4005–4014.
pancreatic beta-cells, Diabetes 56 (2007) 1534–1543.
[15] S. Jacqueminet, I. Briaud, C. Rouault, G. Reach, V. Poitout, Inhibition of insulin
[41] L.L. Listenberger, X. Han, S.E. Lewis, S. Cases, R.V. Farese Jr., D.S. Ory, J.E. Schaffer,
gene expression by long-term exposure of pancreatic beta-cells to palmitate
Triglyceride accumulation protects against fatty acid-induced lipotoxicity, Proc.
is dependent upon the presence of a stimulatory glucose concentration,
Natl. Acad. Sci. U. S. A. 100 (2003) 3077–3082.
Metabolism 49 (2000) 532–536.
[42] A.K. Busch, E. Gurisik, D.V. Cordery, M. Sudlow, G.S. Denyer, D.R. Laybutt, W.E.
[16] I. Briaud, J.S. Harmon, C.L. Kelpe, V.B. Segu, V. Poitout, Lipotoxicity of the
Hughes, T.J. Biden, Increased fatty acid desaturation and enhanced expression of
pancreatic beta-cell is associated with glucose-dependent esterification of fatty
stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapop-
acids into neutral lipids, Diabetes 50 (2001) 315–321.
tosis, Diabetes 54 (2005) 2917–2924.
[17] C.L. Kelpe, P.C. Moore, S.D. Parazzoli, B. Wicksteed, C.J. Rhodes, V. Poitout,
[43] J.M. Ntambi, M. Miyazaki, J.P. Stoehr, H. Lan, C.M. Kendziorski, B.S. Yandell, Y.
Palmitate inhibition of insulin gene expression is mediated at the transcriptional
Song, P. Cohen, J.M. Friedman, A.D. Attie, Loss of stearoyl-CoA desaturase-1
level via ceramide synthesis, J. Biol. Chem. 278 (2003) 30015–30021.
function protects mice against adiposity, Proc. Natl. Acad. Sci. U. S. A. 99 (2002)
[18] D.K. Hagman, L.B. Hays, S.D. Parazzoli, V. Poitout, Palmitate inhibits insulin gene
expression by altering PDX-1 nuclear localization and reducing MafA expression
[44] J.B. Flowers, M.E. Rabaglia, K.L. Schueler, M.T. Flowers, H. Lan, M.P. Keller, J.M.
in isolated rat islets of Langerhans, J. Biol. Chem. 280 (2005) 32413–32418.
Ntambi, A.D. Attie, Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity
[19] A. Pick, J. Clark, C. Kubstrup, M. Levisetti, W. Pugh, S. Bonner-Weir, K. Polonsky,
in lean mice but worsens diabetes in leptin-deficient obese mice, Diabetes 56
Role of apoptosis in failure of beta-cell mass compensation for insulin resistance
(2007) 1228–1239.
and beta-cell defects in the male Zucker diabetes fatty rat, Diabetes 47 (1998)
[45] C.J. Nolan, M. Prentki, The islet beta-cell: fuel responsive and vulnerable, Trends
Endocrinol. Metab. 19 (2008) 285–291.
[20] M. Shimabukuro, Y.-T. Zhou, M. Levi, R.H. Unger, Fatty-acid-induced beta-cell
[46] M. Prentki, S.R. Madiraju, Glycerolipid metabolism and signaling in health and
apoptosis: a link between obesity and diabetes, Proc. Natl. Acad. Sci. U. S. A. 95
disease, Endocr. Rev. 29 (2008) 647–676.
(1998) 2498–2502.
[47] M. Shimabukuro, M. Higa, Y.T. Zhou, M.Y. Wang, C.B. Newgard, R.H. Unger,
[21] M. Cnop, J.C. Hannaert, A. Hoorens, D.L. Eizirik, D.G. Pipeleers, Inverse
Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine
relationship between cytotoxicity of free fatty acids in pancreatic islet cells
palmitoyltransferase overexpression, J. Biol. Chem. 273 (1998) 32487–32490.
and cellular triglyceride accumulation, Diabetes 50 (2001) 1771–1777.
[48] P.C. Moore, M.A. Ugas, D.K. Hagman, S.D. Parazzoli, V. Poitout, Evidence against
[22] K. Maedler, G.A. Spinas, D. Dyntar, W. Moritz, N. Kaiser, M.Y. Donath, Distinct
the involvement of oxidative stress in fatty acid inhibition of insulin secretion,
effects of saturated and monounsaturated fatty acids on beta-cell turnover and
Diabetes 53 (2004) 2610–2616.
function, Diabetes 50 (2001) 69–76.
[49] M. Cnop, J.C. Hannaert, A.Y. Grupping, D.G. Pipeleers, Low density lipoprotein can
[23] R. Lupi, F. Dotta, L. Marselli, S. Del Guerra, M. Masini, C. Santangelo, G. Patane, U.
cause death of islet beta-cells by its cellular uptake and oxidative modification,
Boggi, S. Piro, M. Anello, E. Bergamini, F. Mosca, U. Di Mario, S. Del Prato, P.
Endocrinology 143 (2002) 3449–3453.
Marchetti, Prolonged exposure to free fatty acids has cytostatic and pro-
[50] A. Abderrahmani, G. Niederhauser, D. Favre, S. Abdelli, M. Ferdaoussi, J.Y.
apoptotic effects on human pancreatic islets: evidence that beta-cell death is
Yang, R. Regazzi, C. Widmann, G. Waeber, Human high-density lipoprotein
caspase mediated, partially dependent on ceramide pathway, and Bcl-2
particles prevent activation of the JNK pathway induced by human oxidised
regulated, Diabetes 51 (2002) 1437–1442.
low-density lipoprotein particles in pancreatic beta cells, Diabetologia 50
[24] C.E. Wrede, L.M. Dickson, M.K. Lingohr, I. Briaud, C.J. Rhodes, Protein kinase
(2007) 1304–1314.
B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1),
[51] L.R. Brunham, J.K. Kruit, T.D. Pape, J.M. Timmins, A.Q. Reuwer, Z. Vasanji, B.J.
J. Biol. Chem. 277 (2002) 49676–49684.
Marsh, B. Rodrigues, J.D. Johnson, J.S. Parks, C.B. Verchere, M.R. Hayden, beta-Cell
[25] S. Piro, M. Anello, C. Di Pietro, M.N. Lizzio, G. Patane, A.M. Rabuazzo, R. Vigneri, M.
ABCA1 influences insulin secretion, glucose homeostasis and response to
Purrello, F. Purrello, Chronic exposure to free fatty acids or high glucose induces
thiazolidinedione treatment, Nat. Med. 13 (2007) 340–347.
apoptosis in rat pancreatic islets: possible role of oxidative stress, Metabolism 51
[52] M. Ishikawa, Y. Iwasaki, S. Yatoh, T. Kato, S. Kumadaki, N. Inoue, T. Yamamoto, T.
(2002) 1340–1347.
Matsuzaka, Y. Nakagawa, N. Yahagi, K. Kobayashi, A. Takahashi, N. Yamada, H.
[26] K. Maedler, J. Oberholzer, P. Bucher, G.A. Spinas, M.Y. Donath, Monounsaturated
Shimano, Cholesterol accumulation and diabetes in pancreatic beta-cell-specific
fatty acids prevent the deleterious effects of palmitate and high glucose on
SREBP-2 transgenic mice: a new model for lipotoxicity, J. Lipid Res. 49 (2008)
human pancreatic beta-cell turnover and function, Diabetes 52 (2003) 726–733.
[27] I. Maestre, J. Jordan, S. Calvo, J.A. Reig, V. Cena, B. Soria, M. Prentki, E. Roche,
[53] N. Mitro, P.A. Mak, L. Vargas, C. Godio, E. Hampton, V. Molteni, A. Kreusch, E. Saez,
Mitochondrial dysfunction is involved in apoptosis induced by serum with-
The nuclear receptor LXR is a glucose sensor, Nature 445 (2007) 219–223.
drawal and fatty acids in the beta-cell line INS-1, Endocrinology 144 (2003)
[54] C.P. Briscoe, M. Tadayyon, J.L. Andrews, W.G. Benson, J.K. Chambers, M.M. Eilert,
C. Ellis, N.A. Elshourbagy, A.S. Goetz, D.T. Minnick, P.R. Murdock, H.R. Sauls Jr., U.
[28] W. El-Assaad, J. Buteau, M.L. Peyot, C. Nolan, R. Roduit, S. Hardy, E. Joly, G. Dbaibo,
Shabon, L.D. Spinage, J.C. Strum, P.G. Szekeres, K.B. Tan, J.M. Way, D.M. Ignar, S.
L. Rosenberg, M. Prentki, Saturated fatty acids synergize with elevated glucose to
Wilson, A.I. Muir, The orphan G protein-coupled receptor GPR40 is activated by
cause pancreatic beta-cell death, Endocrinology 144 (2003) 4154–4163.
medium and long chain fatty acids, J. Biol. Chem. 278 (2003) 11303–11311.
[29] J.S. Harmon, C.E. Gleason, Y. Tanaka, V. Poitout, R.P. Robertson, Antecedent
[55] Y. Itoh, Y. Kawamata, M. Harada, M. Kobayashi, R. Fujii, S. Fukusumi, K. Ogi, M.
hyperglycemia, not hyperlipidemia, is associated with increased islet triacylgly-
Hosoya, Y. Tanaka, H. Uejima, H. Tanaka, M. Maruyama, R. Satoh, S. Okubo, H.
cerol content and decreased insulin gene mRNA level in Zucker diabetic fatty
Kizawa, H. Komatsu, F. Matsumura, Y. Noguchi, T. Shinohara, S. Hinuma, Y.
rats, Diabetes 50 (2001) 2481–2486.
Fujisawa, M. Fujino, Free fatty acids regulate insulin secretion from pancreatic
[30] I. Briaud, C.L. Kelpe, L.M. Johnson, P.O.T. Tran, V. Poitout, Differential effects of
beta cells through GPR40, Nature 422 (2003) 173–176.
hyperlipidemia on insulin secretion in islets of Langerhans from hyperglycemic
[56] P. Steneberg, N. Rubins, R. Bartoov-Shifman, M.D. Walker, H. Edlund, TheFFA ,
vs. normoglycemic rats, Diabetes 51 (2002) 662–668.
receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose
[31] M. Prentki, E. Joly, W. El-Assaad, R. Roduit, Malonyl-CoA signaling, lipid
homeostasis in mouse, Cell Metab. 1 (2005) 245–258.
partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in
[57] M.G. Latour, T. Alquier, E. Oseid, C. Tremblay, T.L. Jetton, J. Luo, D.C. Lin, V. Poitout,
the etiology of diabetes, Diabetes 51 (Suppl 3) (2002) S405–S413.
GPR40 is necessary but not sufficient for fatty acid stimulation of insulin
[32] V. Poitout, Lipid partitioning in the pancreatic beta-cell: physiologic and
secretion in vivo, Diabetes 56 (2007) 1087–1094.
pathophysiologic implications, Curr. Opin. Endocrinol. Diabetes 9 (2002)
[58] M. Kebede, T. Alquier, M.G. Latour, M. Semache, C. Tremblay, V. Poitout, The fatty
acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat
[33] T. Brun, E. Roche, F. Assimacopoulos-Jeannet, B.E. Corkey, K.-H. Kim, M. Prentki,
feeding, Diabetes 57 (2008) 2432–2437.
Evidence fore anaplerotic/malonyl-CoA pathway in pancreatic beta-cell nutrient
[59] M. Cnop, M. Igoillo-Esteve, D.A. Cunha, L. Ladriere, D.L. Eizirik, An update on
signaling, Diabetes 45 (1996) 190–198.
lipotoxic endoplasmic reticulum stress in pancreatic beta-cells, Biochem. Soc.
[34] E. Roche, S. Farfari, L.A. Witters, F. Assimacopoulos-Jeannet, S. Thumelin, T. Brun,
Trans. 36 (2008) 909–915.
B.E. Corkey, A.K. Saha, M. Prentki, Long-term exposure of beta-INS cells to high
[60] C.P. Tan, Y. Feng, Y.P. Zhou, G.J. Eiermann, A. Petrov, C. Zhou, S. Lin, G. Salituro, P.
glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene
Meinke, R. Mosley, T.E. Akiyama, M. Einstein, S. Kumar, J.P. Berger, S.G. Mills, N.A.
expression, Diabetes 47 (1998) 1086–1094.
Thornberry, L. Yang, A.D. Howard, Selective small-molecule agonists of G
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
protein-coupled receptor 40 promote glucose-dependent insulin secretion and
[85] C.A. Amezcua, S.M. Harper, J. Rutter, K.H. Gardner, Structure and interactions of
reduce blood glucose in mice, Diabetes 57 (2008) 2211–2219.
PAS kinase N-terminal PAS domain: model for intramolecular kinase regulation,
[61] T.M. Mason, T. Goh, V. Tchipashvili, H. Sandhu, N. Gupta, G.F. Lewis, A. Giacca,
Structure 10 (2002) 1349–1361.
Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory
[86] J. Rutter, B.L. Probst, S.L. McKnight, Coordinate regulation of sugar flux and
response to glucose in vivo in rats, Diabetes 48 (1999) 524–530.
translation by PAS kinase, Cell 111 (2002) 17–28.
[62] G. Paolisso, A. Gambardella, L. Amato, R. Tortoriello, A. D'Amore, M. Varrichio, F.
[87] W.A. Wilson, A.V. Skurat, B. Probst, A. de Paoli-Roach, P.J. Roach, J. Rutter, Control
D'Onofrio, Opposite effects of short- and long-term fatty acid infusion on insulin
of mammalian glycogen synthase by PAS kinase, Proc. Natl. Acad. Sci. U. S. A. 102
secretion in healthy subjects, Diabetologia 38 (1995) 1295–1299.
[63] C.-Y. Zhang, G. Baffy, P. Perret, S. Krauss, O. Peroni, D. Grujic, T. Hagen, A.-J. Vidal-
[88] G. da Silva Xavier, J. Rutter, G.A. Rutter, Involvement of Per-Arnt-Sim (PAS)
Puig, O. Boss, Y.-B. Kim, X.X. Zheng, M.B. Wheeler, G.I. Shulman, C.B. Chan, B.B.
kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox
Lowell, Uncoupling protein-2 negatively regulates insulin secretion and is a
1 gene expression by glucose, Proc. Natl. Acad. Sci. U. S. A. 101 (2004)
major link between obesity, beta-cell dysfunction, and type 2 diabetes, Cell 105
(2001) 745–755.
[89] M. Lu, J. Seufert, J.F. Habener, Pancreatic β-cell-specific repression of insulin gene
[64] J. Nedergaard, D. Ricquier, L.P. Kozak, Uncoupling proteins: current status and
transcription by CCAAT/enhancer-binding protein β. Inhibitory interactions
therapeutic prospects, EMBO Rep. 6 (2005) 917–921.
with basic helix-loop-helix transcription factor E47, J. Biol. Chem. 272 (1997)
[65] C.B. Chan, P.E. MacDonald, M.C. Saleh, D.C. Johns, E. Marban, M.B. Wheeler,
Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin
[90] V. Plaisance, V. Perret, D. Favre, A. Abderrahmani, J.Y. Yang, C. Widmann, R.
secretion from rat islets, Diabetes 48 (1999) 1482–1486.
Regazzi, Role of the transcriptional factor C/EBPbeta in free fatty acid-elicited
[66] C.B. Chan, D. De Leo, J.W. Joseph, T.S. McQuaid, X.F. Ha, F. Xu, R.G. Tsushima, P.S.
beta-cell failure, Mol. Cell. Endocrinol. 305 (2009) 47–55.
Pennefather, A.M. Salapatek, M.B. Wheeler, Increased uncoupling protein-2
[91] M.C. Lawrence, K. McGlynn, B.H. Park, M.H. Cobb, ERK1/2-dependent activation
levels in beta-cells are associated with impaired glucose-stimulated insulin
of transcription factors required for acute and chronic effects of glucose on the
secretion: mechanism of action, Diabetes 50 (2001) 1302–1310.
insulin gene promoter, J. Biol. Chem. 280 (2005) 26751–26759.
[67] J.W. Joseph, V. Koshkin, C.Y. Zhang, J. Wang, B.B. Lowell, C.B. Chan, M.B. Wheeler,
[92] D.L. Eizirik, A.K. Cardozo, M. Cnop, The role for endoplasmic reticulum stress in
Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity
diabetes mellitus, Endocr. Rev. 29 (2008) 42–61.
after a high-fat diet, Diabetes 51 (2002) 3211–3219.
[93] D.R. Laybutt, A.M. Preston, M.C. Akerfeldt, J.G. Kench, A.K. Busch, A.V. Biankin, T.J.
[68] J. Pi, Y. Bai, K.W. Daniel, D. Liu, O. Lyght, D. Edelstein, M. Brownlee, B.E. Corkey, S.
Biden, Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2
Collins, Persistent oxidative stress due to absence of uncoupling protein 2
diabetes, Diabetologia 50 (2007) 752–763.
associated with impaired pancreatic beta-cell function, Endocrinology 7 (2009)
[94] E. Karaskov, C. Scott, L. Zhang, T. Teodoro, M. Ravazzola, A. Volchuk, Chronic
palmitate but not oleate exposure induces endoplasmic reticulum stress, which
[69] N. Lameloise, P. Muzzin, M. Prentki, F. Assimacopoulos-Jeannet, Uncoupling
may contribute to INS-1 pancreatic beta-cell apoptosis, Endocrinology 147
protein 2: a possible link between fatty acid excess and impaired glucose-
(2006) 3398–3407.
induced insulin secretion? Diabetes 50 (2001) 803–809.
[95] M. Cnop, L. Ladriere, P. Hekerman, F. Ortis, A.K. Cardozo, Z. Dogusan, D. Flamez,
[70] G. Patane, M. Anello, S. Piro, R. Vigneri, F. Purrello, A.M. Rabuazzo, Role of ATP
M. Boyce, J. Yuan, D.L. Eizirik, Selective inhibition of eukaryotic translation
production and uncoupling protein-2 in the insulin secretory defect induced by
initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced
chronic exposure to high glucose or free fatty acids and effects of peroxisome
endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and
proliferator-activated receptor-gamma inhibition, Diabetes 51 (2002) 2749–2756.
apoptosis, J. Biol. Chem. 282 (2007) 3989–3997.
[71] N. Produit-Zengaffinen, N. Davis-Lameloise, H. Perreten, D. Becard, A. Gjinovci, P.A.
[96] H. Wang, G. Kouri, C.B. Wollheim, ER stress and SREBP-1 activation are
Keller, C.B. Wollheim, P. Herrera, P. Muzzin, F. Assimacopoulos-Jeannet, Increasing
implicated in beta-cell glucolipotoxicity, J. Cell Sci. 118 (2005) 3905–3915.
uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin
[97] I. Kharroubi, L. Ladriere, A.K. Cardozo, Z. Dogusan, M. Cnop, D.L. Eizirik, Free fatty
secretion but decreases production of reactive oxygen species, Diabetologia 50
acids and cytokines induce pancreatic beta-cell apoptosis by different mechan-
(2007) 84–93.
isms: role of nuclear factor-kappaB and endoplasmic reticulum stress,
[72] C. Schmitz-Peiffer, D.R. Laybutt, J.G. Burchfield, E. Gurisik, S. Narasimhan, C.J.
Endocrinology 145 (2004) 5087–5096.
Mitchell, D.J. Pedersen, U. Braun, G.J. Cooney, M. Leitges, T.J. Biden, Inhibition of
[98] S.C. Martinez, K. Tanabe, C. Cras-Meneur, N.A. Abumrad, E. Bernal-Mizrachi, M.A.
PKCepsilon improves glucose-stimulated insulin secretion and reduces insulin
Permutt, Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid
clearance, Cell Metab. 6 (2007) 320–328.
and endoplasmic reticulum stress-induced apoptosis, Diabetes 57 (2008)
[73] J. Cantley, J.G. Burchfield, G.L. Pearson, C. Schmitz-Peiffer, M. Leitges, T.J. Biden,
Deletion of PKCepsilon selectively enhances the amplifying pathways of glucose-
[99] D.A. Cunha, P. Hekerman, L. Ladriere, A. Bazarra-Castro, F. Ortis, M.C. Wakeham,
stimulated insulin secretion via increased lipolysis in mouse beta-cells, Diabetes
F. Moore, J. Rasschaert, A.K. Cardozo, E. Bellomo, L. Overbergh, C. Mathieu, R.
58 (2009) 1826–1834.
Lupi, T. Hai, A. Herchuelz, P. Marchetti, G.A. Rutter, D.L. Eizirik, M. Cnop, Initiation
[74] M.L. Peyot, C. Guay, M.G. Latour, J. Lamontagne, R. Lussier, M. Pineda, N.B.
and execution of lipotoxic ER stress in pancreatic beta-cells, J. Cell Sci. 121
Ruderman, G. Haemmerle, R. Zechner, E. Joly, S.R. Madiraju, V. Poitout, M.
(2008) 2308–2318.
Prentki, Adipose triglyceride lipase is implicated in fuel- and non-fuel-
[100] E. Bachar, Y. Ariav, M. Ketzinel-Gilad, E. Cerasi, N. Kaiser, G. Leibowitz, Glucose
stimulated insulin secretion, J. Biol. Chem. 284 (2009) 16848–16859.
amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-
[75] T. Kato, H. Shimano, T. Yamamoto, T. Yokoo, Y. Endo, M. Ishikawa, T. Matsuzaka,
cells via activation of mTORC1, PLoS One 4 (2009) e4954.
Y. Nakagawa, S. Kumadaki, N. Yahagi, A. Takahashi, H. Sone, H. Suzuki, H.
[101] K.S. Gwiazda, T.L. Yang, Y. Lin, J.D. Johnson, Effects of palmitate on ER and
Toyoshima, A.H. Hasty, S. Takahashi, H. Gomi, T. Izumi, N. Yamada, Granuphilin is
cytosolic Ca2+ homeostasis in beta-cells, Am. J. Physiol., Endocrinol. Metab. 296
activated by SREBP-1c and involved in impaired insulin secretion in diabetic
(2009) E690–701.
mice, Cell Metab. 4 (2006) 143–154.
[102] P.C. Moore, D.C. Lin, J. Luo, V. Poitout, Deletion of the GPR40 gene impairs fatty-
[76] C.S. Olofsson, S. Collins, M. Bengtsson, L. Eliasson, A. Salehi, K. Shimomura, A.
acid potentiation of insulin secretion in isolated mouse islets, Diabetes 54
Tarasov, C. Holm, F. Ashcroft, P. Rorsman, Long-term exposure to glucose and
(Suppl.1) (2005) A83.
lipids inhibits glucose-induced insulin secretion downstream of granule fusion
[103] H.Y. Seo, Y.D. Kim, K.M. Lee, A.K. Min, M.K. Kim, H.S. Kim, K.C. Won, J.Y. Park, K.U.
with plasma membrane, Diabetes 56 (2007) 1888–1897.
Lee, H.S. Choi, K.G. Park, I.K. Lee, Endoplasmic reticulum stress-induced
[77] D.K. Hagman, M.G. Latour, S.K. Chakrabarti, G. Fontes, J. Amyot, C. Tremblay, M.
activation of activating transcription factor 6 decreases insulin gene expression
Semache, J.A. Lausier, V. Roskens, R.G. Mirmira, T.L. Jetton, V. Poitout, Cyclical and
via up-regulation of orphan nuclear receptor small heterodimer partner,
alternating infusions of glucose and intralipid in rats inhibit insulin gene
Endocrinology 149 (2008) 3832–3841.
expression and Pdx-1 binding in islets, Diabetes 57 (2008) 424–431.
[104] C. Evans-Molina, R.D. Robbins, T. Kono, S.A. Tersey, G.L. Vestermark, C.S.
[78] J.S. Harmon, R. Stein, R.P. Robertson, Oxidative stress-mediated, post-transla-
Nunemaker, J.C. Garmey, T.G. Deering, S.R. Keller, B. Maier, R.G. Mirmira,
tional loss of MafA protein as a contributing mechanism to loss of insulin gene
Peroxisome proliferator-activated receptor gamma activation restores islet
expression in glucotoxic beta cells, J. Biol. Chem. 280 (2005) 11107–11113.
function in diabetic mice through reduction of endoplasmic reticulum stress and
[79] S. Mathias, L.A. Pena, R.N. Kolesnick, Signal transduction of stress via ceramide,
maintenance of euchromatin structure, Mol. Cell. Biol. 29 (2009) 2053–2067.
Biochem. J. 335 (Pt. 3) (1998) 465–480.
[105] P. Pirot, N. Naamane, F. Libert, N.E. Magnusson, T.F. Orntoft, A.K. Cardozo, D.L.
[80] E. Henderson, R. Stein, c-jun inhibits transcriptional activation by the insulin
Eizirik, Global profiling of genes modified by endoplasmic reticulum stress in
enhancer, and the insulin control element is the target of control, Mol. Cell. Biol.
pancreatic beta cells reveals the early degradation of insulin mRNAs, Diabetologia
14 (1994) 655–662.
50 (2007) 1006–1014.
[81] G.L. Robinson, E. Henderson, M.E. Massari, C. Murre, R. Stein, c-jun inhibits
[106] X. Wang, H. Li, D. De Leo, W. Guo, V. Koshkin, I.G. Fantus, A. Giacca, C.B. Chan, S.
insulin control element-mediated transcription by affecting the transactivation
Der, M.B. Wheeler, Gene and protein kinase expression profiling of reactive
potential of the E2A gene products, Mol. Cell. Biol. 15 (1995) 1398–1404.
oxygen species-associated lipotoxicity in the pancreatic beta-cell line MIN6,
[82] H. Kaneto, G. Xu, N. Fujii, S. Kim, S. Bonner-Weir, G.C. Weir, Involvement of c-Jun
Diabetes 53 (2004) 129–140.
N-terminal kinase in oxidative stress-mediated suppression of insulin gene
[107] D. Morgan, H.R. Oliveira-Emilio, D. Keane, A.E. Hirata, M. Santos da Rocha, S.
expression, J. Biol. Chem. 277 (2002) 30010–30018.
Bordin, R. Curi, P. Newsholme, A.R. Carpinelli, Glucose, palmitate and pro-
[83] G. Solinas, W. Naugler, F. Galimi, M.S. Lee, M. Karin, Saturated fatty acids inhibit
inflammatory cytokines modulate production and activity of a phagocyte-like
induction of insulin gene transcription by JNK-mediated phosphorylation of
NADPH oxidase in rat pancreatic islets and a clonal beta cell line, Diabetologia 50
insulin-receptor substrates, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 16454–16459.
(2007) 359–369.
[84] G. Fontés, M. Semache, D.K. Hagman, C. Tremblay, R. Shah, C.J. Rhodes, J. Rutter,
[108] A.K. Busch, D. Cordery, G.S. Denyer, T.J. Biden, Expression profiling of palmitate-
V. Poitout, Involvement of PAS Kinase and ERK1/2 in Palmitate Inhibition of
and oleate-regulated genes provides novel insights into the effects of chronic
Insulin Gene Expression In Pancreatic Beta-Cells, Diabetes 58 (2009) 2048–2058.
lipid exposure on pancreatic beta-cell function, Diabetes 51 (2002) 977–987.
Author's personal copy
V. Poitout et al. / Biochimica et Biophysica Acta 1801 (2010) 289–298
[109] K.D. Jeffrey, E.U. Alejandro, D.S. Luciani, T.B. Kalynyak, X. Hu, H. Li, Y. Lin, R.R.
activity and leads to increased beta-cell responsiveness to glucose, J. Clin. Invest.
Townsend, K.S. Polonsky, J.D. Johnson, Carboxypeptidase E mediates palmitate-
103 (1999) 413–419.
induced beta-cell ER stress and apoptosis, Proc. Natl. Acad. Sci. U. S. A. 105 (2008)
[127] G.M. Steil, N. Trivedi, J.C. Jonas, W.M. Hasenkamp, A. Sharma, S. Bonner-Weir,
G.C. Weir, Adaptation of beta-cell mass to substrate oversupply: enhanced
[110] E. Lai, G. Bikopoulos, M.B. Wheeler, M. Rozakis-Adcock, A. Volchuk, Differential
function with normal gene expression, Am. J. Physiol., Endocrinol. Metab. 280
activation of ER stress and apoptosis in response to chronically elevated free
(2001) E788–796.
fatty acids in pancreatic beta-cells, Am. J. Physiol., Endocrinol. Metab. 294 (2008)
[128] T.T. Goh, T.M. Mason, N. Gupta, A. So, T.K. Lam, L. Lam, G.F. Lewis, A. Mari, A.
Giacca, Lipid-induced beta-cell dysfunction in vivo in models of progressive
[111] C.J. Huang, C.Y. Lin, L. Haataja, T. Gurlo, A.E. Butler, R.A. Rizza, P.C. Butler, High
beta-cell failure, Am. J. Physiol., Endocrinol. Metab. 292 (2007) E549–E560.
expression rates of human islet amyloid polypeptide induce endoplasmic
[129] A.I. Oprescu, G. Bikopoulos, A. Naassan, E.M. Allister, C. Tang, E. Park, H. Uchino,
reticulum stress mediated beta-cell apoptosis, a characteristic of humans with
G.F. Lewis, I.G. Fantus, M. Rozakis-Adcock, M.B. Wheeler, A. Giacca, Free fatty
type 2 but not type 1 diabetes, Diabetes 56 (2007) 2016–2027.
acid-induced reduction in glucose-stimulated insulin secretion: evidence for a
[112] W.F. Graier, R. Malli, G.M. Kostner, Mitochondrial protein phosphorylation:
role of oxidative stress in vitro and in vivo, Diabetes 56 (2007) 2927–2937.
instigator or target of lipotoxicity? Trends. Endocrinol. Metab. 20 (2009) 186–193.
[130] M.M. Rankin, J.A. Kushner, Adaptive beta-cell proliferation is severely restricted
[113] V. Koshkin, F.F. Dai, C.A. Robson-Doucette, C.B. Chan, M.B. Wheeler, Limited
with advanced age, Diabetes 58 (2009) 1365–1372.
mitochondrial permeabilization is an early manifestation of palmitate-induced
[131] S.I. Tschen, S. Dhawan, T. Gurlo, A. Bhushan, Age-dependent decline in beta-cell
lipotoxicity in pancreatic beta-cells, J. Biol. Chem. 283 (2008) 7936–7948.
proliferation restricts the capacity of beta-cell regeneration in mice, Diabetes 58
[114] D.S. Luciani, K.S. Gwiazda, T.L. Yang, T.B. Kalynyak, Y. Bychkivska, M.H. Frey, K.D.
(2009) 1312–1320.
Jeffrey, A.V. Sampaio, T.M. Underhill, J.D. Johnson, Roles of IP3R and RyR Ca2+
[132] G. Boden, X. Chen, J. Rosner, M. Barton, Effects of a 48h fat infusion on insulin
channels in endoplasmic reticulum stress and beta-cell death, Diabetes 58
secretion and glucose utilization, Diabetes 44 (1995) 1239–1242.
(2009) 422–432.
[133] G. Boden, X. Chen, Effects of fatty acids and ketone bodies on basal insulin
[115] P. Lovis, E. Roggli, D.R. Laybutt, S. Gattesco, J.Y. Yang, C. Widmann, A.
secretion in type 2 diabetes, Diabetes 48 (1999) 577–583.
Abderrahmani, R. Regazzi, Alterations in microRNA expression contribute to
[134] A. Carpentier, S. Mittelman, B. Lamarche, R. Bergman, A. Giacca, G. Lewis, Acute
fatty acid-induced pancreatic beta-cell dysfunction, Diabetes 57 (2008)
enhancement of insulin secretion by FFA in humans is lost with prolonged FFA
elevation, Am. J. Physiol. (1999) 276.
[116] G.V. Richieri, A. Anel, A.M. Kleinfeld, Interactions of long-chain fatty acids and
[135] A. Carpentier, A. Giacca, G.F. Lewis, Effect of increased plasma non-esterified fatty
albumin: determination of free fatty acid levels using the fluorescent probe
acids (NEFAs) on arginine-stimulated insulin secretion in obese humans,
ADIFAB, Biochemistry 32 (1993) 7574–7580.
Diabetologia 44 (2001) 1989–1997.
[117] G.V. Richieri, A.M. Kleinfeld, Unbound free fatty acid levels in human serum,
[136] A. Carpentier, S.D. Mittelman, R.N. Bergman, A. Giacca, G.F. Lewis, Prolonged
J. Lipid Res. 36 (1995) 229–240.
elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese
[118] W.S. Cruz, G. Kwon, C.A. Marshall, M.L. McDaniel, C.F. Semenkovich, Glucose and
nondiabetic humans but not in individuals with type 2 diabetes, Diabetes 49
insulin stimulate heparin-releasable lipoprotein lipase activity in mouse islets
(2000) 399–408.
and INS-1 cells. A potential link between insulin resistance and beta-cell
[137] N. Leung, T. Sakaue, A. Carpentier, K. Uffelman, A. Giacca, G.F. Lewis, Prolonged
dysfunction, J. Biol. Chem. 276 (2001) 12162–12168.
increase of plasma non-esterified fatty acids fully abolishes the stimulatory
[119] K.L. Pappan, Z. Pan, G. Kwon, C.A. Marshall, T. Coleman, I.J. Goldberg, M.L.
effect of 24 hours of moderate hyperglycaemia on insulin sensitivity and
McDaniel, C.F. Semenkovich, Pancreatic beta-cell lipoprotein lipase indepen-
pancreatic beta-cell function in obese men, Diabetologia 47 (2004) 204–213.
dently regulates islet glucose metabolism and normal insulin secretion, J. Biol.
[138] C. Xiao, A. Giacca, A. Carpentier, G.F. Lewis, Differential effects of monounsat-
Chem. 280 (2005) 9023–9029.
urated, polyunsaturated and saturated fat ingestion on glucose-stimulated
[120] R.H. Unger, Minireview: weapons of lean body mass destruction: the role of
insulin secretion, sensitivity and clearance in overweight and obese, non-
ectopic lipids in the metabolic syndrome, Endocrinology 144 (2003) 5159–5165.
diabetic humans, Diabetologia 49 (2006) 1371–1379.
[121] Y. Lee, H. Hirose, M. Ohneda, J.H. Johnson, J.D. McGarry, R.H. Unger, Beta-cell
[139] C. Xiao, A. Giacca, G.F. Lewis, Oral taurine but not N-acetylcysteine ameliorates
lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of
NEFA-induced impairment in insulin sensitivity and beta cell function in obese
obese rats: impairment in adipocyte–beta-cell relationships, Proc. Natl. Acad. Sci.
and overweight, non-diabetic men, Diabetologia 51 (2008) 139–146.
U. S. A. 91 (1994) 10878–10882.
[140] S. Kashyap, R. Belfort, A. Gastaldelli, T. Pratipanawatr, R. Berria, W. Pratipanawatr,
[122] J.M. Aerts, R. Ottenhoff, A.S. Powlson, A. Grefhorst, M. van Eijk, P.F. Dubbelhuis, J.
M. Bajaj, L. Mandarino, R. DeFronzo, K. Cusi, A sustained increase in plasma free
Aten, F. Kuipers, M.J. Serlie, T. Wennekes, J.K. Sethi, S. O'Rahilly, H.S. Overkleeft,
fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed
Pharmacological inhibition of glucosylceramide synthase enhances insulin
to develop type 2 diabetes, Diabetes 52 (2003) 2461–2474.
sensitivity, Diabetes 56 (2007) 1341–1349.
[141] K. Cusi, S. Kashyap, A. Gastaldelli, M. Bajaj, E. Cersosimo, Effects on insulin
[123] W.L. Holland, J.T. Brozinick, L.P. Wang, E.D. Hawkins, K.M. Sargent, Y. Liu, K.
secretion and insulin action of a 48-h reduction of plasma free fatty acids with
Narra, K.L. Hoehn, T.A. Knotts, A. Siesky, D.H. Nelson, S.K. Karathanasis, G.K.
Acipimox in nondiabetic subjects genetically predisposed to type 2 diabetes, Am.
Fontenot, M.J. Birnbaum, S.A. Summers, Inhibition of ceramide synthesis
J. Physiol., Endocrinol. Metab. 292 (2007) E1775–E1781.
ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resis-
[142] M. Fex, M.D. Nitert, N. Wierup, F. Sundler, C. Ling, H. Mulder, Enhanced
tance, Cell Metab. 5 (2007) 167–179.
mitochondrial metabolism may account for the adaptation to insulin resistance
[124] H. Zhao, M. Przybylska, I.H. Wu, J. Zhang, C. Siegel, S. Komarnitsky, N.S. Yew, S.H.
in islets from C57BL/6J mice fed a high-fat diet, Diabetologia 50 (2006) 74–83.
Cheng, Inhibiting glycosphingolipid synthesis improves glycemic control and
[143] C.J. Nolan, J.L. Leahy, V. Delghingaro-Augusto, J. Moibi, K. Soni, M.L. Peyot, M.
insulin sensitivity in animal models of type 2 diabetes, Diabetes 56 (2007)
Fortier, C. Guay, J. Lamontagne, A. Barbeau, E. Przybytkowski, E. Joly, P. Masiello,
S. Wang, G.A. Mitchell, M. Prentki, Beta-cell compensation for insulin resistance
[125] D.T. Stein, B.E. Stevenson, M.W. Chester, M. Basit, M.B. Daniels, S.D. Turley, J.D.
in Zucker fatty rats: increased lipolysis and fatty acid signalling, Diabetologia 49
McGarry, The insulinotropic potency of fatty acids is influenced profoundly by
(2006) 2120–2130.
their chain length and degree of saturation, J. Clin. Invest. 100 (1997) 398–403.
[144] G. Leibowitz, G. Uckaya, A.I. Oprescu, E. Cerasi, D.J. Gross, N. Kaiser, Glucose-
[126] C. Magnan, S. Collins, M.F. Berthault, N. Kassis, M. Vincent, M. Gilbert, L. Penicaud,
regulated proinsulin gene expression is required for adequate insulin production
A. Ktorza, F. Assimacopoulos-Jeannet, Lipid infusion lowers sympathetic nervous
during chronic glucose exposure, Endocrinology 143 (2002) 3214–3220.
Source: http://poitoutlab.ca/Publications/Published%20article-VP.pdf
15-daagse rondreis israël
15-tägige Rundreise nach Jordanien 19. Mai - 2. Juni 2014 3 Meere – 3 Wüsten – 3 Städte. Höhepunkte: 7 Offenbarungsberge Gottes – Tiefstpunkt der Erde: Das Tote Meer Israel, das Land in dem Gott Geschichte gemacht hat. Wir besuchen berühmte Städte wie Tel
Microsoft word - als_package.revisedfeb2010[1].doc
SOUTH WEST HEALTHCARE WARRNAMBOOL ADVANCED LIFE SUPPORT LEARNING PACKAGE Prepared by: J. Brown, ICU Last revised February, 2010. C. Joseph, Nursing Education. CONTENTS: Basic Life Support Choking and airway obstruction Defibrillation in ALS Automated External Defibrillators