Microsoft word - screening neonatale. cart audiologia
La sordità infantile: lo screening uditivo neonatale universa- le, il percorso del paziente ipoacusico in età pediatrica. Alessandro Martinia, Paola Marchisiob, Luciano Bubbicoc, Patrizia Trevisia, Lodovico Perlettid a Dip. Neuroscienze e Organi di Senso – UOC Otochirurgia e Cattedra di Audiologia, Uni- versità di Padova b Professore Associato di Pediatria, Fondazione Ca' Granda Ospedale Maggiore Policlinico, c Dip. Scienze Biomediche-Istituto italiano di medicina sociale, Roma d Pediatra, Componente del a Commissione nazionale per i Livel i essenziali di Assistenza del Ministero del a salute, Milano Le più recenti acquisizioni scientifiche e tecnologiche hanno consentito di modificare, in modo molto favorevole, la storia naturale della sordità infantile. Lo screening uditivo ne- onatale universale con le otoemissioni permette di anticipare la diagnosi e di iniziare rapi- damente il programma di riabilitazione che, nei casi di sordità grave o profonda, può ri- chiedere l'impianto cocleare. Vengono affrontati gli aspetti organizzativi e gestionali degli interventi nel campo del a sordità infantile, che ci vedono ancora in ritardo rispetto ad altri paesi europei, e in particolare le problematiche che più direttamente coinvolgono i pediatri- neonatologi ospedalieri e di famiglia. Seguendo le raccomandazioni del 'American Academy of Pediatrics, dello European Consensus Statement on Neonatal Hearing Screening e del Joint Committee on Infant Hearing(1-5), lo screening uditivo neonatale universale, attraverso la registrazione delle o- toemissioni, si è diffuso, non solo negli U.S.A., ma ormai in una gran parte dei paesi eu- ropei. In Italia, nel marzo del 2007, è stato inserito nella proposta di revisione dei livel i es- senziali di assistenza (LEA). Purtroppo le difficoltà economiche e la mancata definizione delle priorità nell'ambito del pacchetto dei nuovi LEA, hanno impedito il definitivo inseri- mento del o screening neonatale universale fra le prestazioni previste nel 'ambito del Ser- vizio sanitario nazionale. Nel nostro paese solo il 60% circa dei neonati effettua lo screening, spesso con interruzioni legate al a cronica carenza di personale e apparecchia- Con lo scopo di definire una chiara Linea di indirizzo per l'esecuzione del o screening uditivo neonatale universale, la definizione del percorso del paziente ipoacusico e le indi- cazioni al 'impianto cocleare, il Dipartimento programmazione del Ministero del a salute nominava nel 2010 una commissione con la col aborazione delle principali società scienti- fiche coinvolte. Alcuni autori (AM, PM e LP) del presente lavoro, che hanno partecipato al- la elaborazione del documento ministeriale, hanno ritenuto di portare un contributo al te- ma, visti i ritardi nel a programmazione sanitaria nazionale e regionale e la necessità di un confronto con le esperienze di altri paesi europei. Questo contributo è indirizzato ai pedia- tri e ai neonatologi ospedalieri, ai pediatri di famiglia, agli audiologi e foniatri e a tutto il personale di area pediatrica che si occupa e si occuperà del a diagnosi, cura e riabilitazio-ne del a sordità infantile, sia dal punto di vista sanitario che sociale. Se ne raccomanda una attenta lettura anche agli amministratori ed a chi si occupa di politiche sanitarie nel nostro paese, in quanto lo screening audiologico neonatale costituisce uno degli interventi sanitari con un più favorevole rapporto costi/benefici. 1. LA SORDITÀ INFANTILE1.1 La necessità di una diagnosi precoce
Una normale funzione uditiva è il requisito indispensabile per un regolare sviluppo del linguaggio, caratterizzato da una precisa e progressiva strutturazione del sistema uditivo nervoso centrale e dei sistemi di ideazione e articolazione fonatoria. Inizia a partire dai primi mesi di vita, mantiene la sua criticità nei primi 2-3 anni di vita e si considera esaurito In questo periodo avviene lo sviluppo e l'armonizzazione plastica dei sistemi di elabo- razione, memorizzazione ed apprendimento cerebrale, in particolare si realizza un consi- derevole aumento del e popolazioni neuronali e la loro organizzazione attraverso connes- sioni interneuronali. Tali connessioni rappresentano i circuiti cerebrali deputati al a realiz- zazione di importanti funzioni cerebrali superiori tra le quali il linguaggio. Nel bambino, per potersi sviluppare una buona acquisizione linguistica, deve essere integro il sistema uditivo ed essere sol ecitato da un'adeguata stimolazione sonora in gra- do di attivare i sistemi di integrazione cerebrale. Se la sordità viene rilevata in ritardo e la stimolazione acustica non avviene entro i primi mesi di vita, la sollecitazione delle vie uditi- ve, del e aree associative e dei circuiti cerebrali del SNC non verrà effettuata adeguata-mente, e l'acquisizione del linguaggio verrà compromessa in modo irreversibile. L'età maggiormente critica nel o sviluppo del linguaggio è rappresentata dai primi 2 anni di vita, ma soprattutto risultano essere cruciali i primi 6-8 mesi di vita (6-7-8). Un danno del sistema uditivo avvenuto in questo periodo tende ad alterare i sistemi di elaborazione, di apprendimento, di memorizzazione e di sviluppo cognitivo, riducendo la funzione dei si- stemi di integrazione centrale del e informazioni. L'handicap uditivo insorto in età infantile, oltre ad essere causa di ritardo nell'acquisizione del a competenza linguistica e comunicativa, incide sul a sfera cognitiva, emotivo-affettiva e sociale del bambino. Spesso il bambino sordo tende a sviluppare un linguaggio incomprensibile e viene isolato socialmente dal mondo degli udenti. L'American Academy of Pediatrics e l'European Consensus Statement on Neonatal Hearing Screening, hanno dedicato grande attenzione al a diagnosi precoce del a sordità infantile, fissando l'obiettivo di identificare e avviare a una terapia riabilitativa (protesizza- zione acustica e logopedia) entro i 6 mesi di età (1-5). È dimostrato che ciò è possibile solo sottoponendo a screening tutta la popolazione di neonati. Lo screening del a sola popola- zione a rischio audiologico (Tabel a 1) comporterebbe il mancato riconoscimento di un numero molto elevato di bambini con sordità congenita (circa il 30-40% dei casi). Numero- si studi hanno dimostrato che senza uno screening al a nascita la diagnosi di sordità con- genita viene effettuata troppo tardi(9-12); un'indagine condotta in Belgio(13) ha posto in evi- denza tempi di diagnosi molto lunghi: per le sordità profonde una mediana di 13 mesi, per le severe 27 mesi, ben 45 mesi per le sordità modeste. Negli USA il sistema di sorveglian- za nazionale(6,14) ha rivelato che l'età media del a conferma di diagnosi, in soggetti sotto- posti al o screening neonatale, rispetto ai non screenati era pari a 2,0 mesi rispetto ai 15,0 mesi per le forme di sordità severa e profonda e di 4,0 mesi rispetto ai 25,0 mesi per le forme lievo-moderate. Considerando le gravi conseguenze che a lungo termine la sordità provoca nella per- sona affetta, si ritiene importante sostenere i costi sanitari di uno screening neonatale uni- versale che presenta un ottimo rapporto costi/benefici. 2. EPIDEMIOLOGIA
2.1 Prevalenza
La prevalenza del a sordità neonatale può essere stimata nei paesi occidentali fra 0,5 e 1,5 casi ogni mil e nati, essa rappresenta il difetto sensitivo più frequente nei neonati ed una del e più comuni anomalie congenite. È inoltre evidente l'importanza del 'entità del- la perdita uditiva, in quanto questa condiziona l'intervento riabilitativo e l'eventuale impian- to cocleare. Per i bambini al di sotto dei 15 anni l'Organizzazione Mondiale del a Sanità ha fissato una perdita uditiva permanente (media per le frequenza 0,5-4 kHz) di oltre 31 dB HL nel 'orecchio migliore (BEHL: "better ear hearing level") come limite oltre il quale è rac- comandabile una protesizzazione acustica, rispetto ad oltre 41 dB HL per gli adulti. Studi altamente rappresentativi del a situazione europea sono stati condotti nel Re- gno Unito(15-17), in particolare va segnalato quel o condotto nel a regione inglese di Trent (4,8 milioni di abitanti), su coorti degli anni 1985-1993. Considerando le ipoacusie conge- nite (=>40 dB) la prevalenza risulta del 1,12‰, mentre aggiungendo le ipoacusie acquisite e ad esordio ritardato il tasso aumenta a 1,33‰. Suddividendo le forme congenite per classi di sordità, la prevalenza è di 0,64‰ per perdite fra 40 e 69 dB, di 0,23‰ per perdite fra 70 e 94 dB, e di 0,24‰ per perdite oltre 94 dB, mancano però le forme lievi. Negli USA un deficit uditivo definito come una ipoacusia bilaterale neurosensoriale pari o superiore a 35 dB è stato calcolato essere, al a nascita, pari a 1,86‰. La prevalen- za continua ad aumentare con l'età, raggiungendo i 2,7‰ a 5 anni e il 3,5‰ durante l'adolescenza(18). Molti sono gli studi condotti in Italia, in particolare dal gruppo di Ferrara (19-24), Un recente studio italiano(25) ha dimostrato con uno screening neonatale, condotto su 19.700 neonati, che la prevalenza delle sordità congenite era pari al '1,78‰, le forme di sordità bilaterale erano presenti nel '1,45‰; le forme lievi, con una perdita pari a 30 dB, erano il 2,9%, quel e con perdita lieve-media fra 30-50 dB erano il 5,7%, le forme medio- severe fra 50-80 dB erano il 25,7% e quel e con perdita severa, 80-100 dB, erano il Una ricerca retrospettiva condotta in Italia dal 'Istituto Italiano di Medicina Sociale su un campione di 40.887 casi con perdita del a capacità uditiva neuro-sensoriale profonda prelinguale presenti in Italia nel 2003, riconosciuti come sordomuti dal a Commissione prevista dal a legge 508, Art. 4 del 1988, ha dimostrato una prevalenza di 0,72 per 1000 Esistono significative differenze su base regionale, le regioni a più elevata prevalen- za di sordità prelinguale profonda sono la Basilicata (1,17‰), la Calabria (1,08‰) e la Si- cilia (1,25‰). Pur con i limiti legati ad una analisi retrospettiva, i tassi di prevalenza delle sordità congenite sembrano diminuire gradualmente nel tempo, questo può essere spiegato con la migliore attenzione al e malattie infettive in gravidanza e in generale al a salute del a donna in età fertile. In particolare si è osservata una nettissima diminuzione del a rosolia materna, una del e principali cause eziologiche della sordità congenita, dopo l'introduzione del a vaccinazione antirubeolica. L'alta percentuale, fino a 7-10 volte il dato nazionale, in alcune comunità rurali del e regioni del Sud può essere molto probabilmente legata a fattori genetici per matrimoni fra consanguinei. 3. EZIOLOGIA
3.1 Generalità
Fra le ipoacusie permanenti del 'infanzia, si pensa che circa il 50-60% possano esse- re costituite da forme genetiche (probabilmente la maggior parte di forme ad "eziologia sconosciuta" rientrano in questa categoria). La restante quota, attorno al 40-50% è costi- tuita da forme acquisite. In circa il 30% dei casi di sordità infantile sono presenti altri di-sordini che interessano differenti organi ed apparati: cranio-facciali, muscolo-scheletrici, cardio-vascolari, oculari, del sistema nervoso centrale, ed altri(26). Attorno al 30% viene stimata l'occorrenza di forme di ipoacusia infantile ad andamento progressivo (Figura 1). Una analisi condotta su 87.000 neonati, in un model o organizzativo che prevedeva un'attenta ricerca dei fattori eziologici(27), dimostra che una causa può essere identificata in oltre la metà dei soggetti con sordità congenita (55,2%); fattori genetici erano presenti nel 33% del totale, nell'11% le infezioni, di cui circa il 10% dovute al Citomegalovirus, le cause perinatali rappresentavano il 10% (Figura 2). 3.2 Sordità genetiche
La maggioranza del e ipoacusie genetiche ereditarie prelinguali non sindromiche (80%) sono trasmesse con modalità autosomica recessiva, sono di solito gravi-profonde e stabili nel tempo(28-31). Le più studiate sono quel e relative al e mutazioni nei geni che codi- ficano per le connessine (CX) 26 e 30 rispettivamente GJB2 e GJB6 (32-37). Le connessine sono proteine necessarie per gli scambi intercel ulari; la mancata sintesi del e connessine determinerebbe ipoacusia da intossicazione da K del 'organo del Corti per mancato riassor- bimento di ioni K+ dal e sinapsi al a base del e cel ule ciliate. Una quota inferiore di ipoacusie ereditarie non sindromiche (15% in età preverbale) è trasmessa con modalità autosomica dominante. Generalmente queste forme causano una ipoacusia meno grave del e forme recessive, spesso postlinguale, con un andamento progressivo nel tempo, per cui sono le forme più difficili da riconoscere nel a popolazione infantile. La restante quota di sordità ereditarie non sindromiche (2-3%) comprende quel e trasmesse con modalità "X-linked" e quel e associate ad una mutazione del DNA mito- condriale (38-41). Fra le ipoacusie genetiche sindromiche (circa 500 tipi), la sindrome di Alport (disturbi renali, oculari e cocleari), la sindrome di Jervel e Lange-Nielsen (ipoacusia grave e al un- gamento del tratto Q-T), la sindrome di Treacher-Col ins (ipoaplasia del e ossa zigomati- che e del a mandibola, coloboma), la sindrome di Usher (retinopatia associata), la sin- drome di Pendred (gozzo), la sindrome di Waardenburg (anomalie di pigmentazione) co- stituiscono esempi in cui la ipoacusia è associata a disordini multi-sistemici che richiedono trattamenti riabilitativi complessi. (41-50) 3.3 Cause perinatali
Le più frequenti cause perinatali sono costituite da:
a) Prematurità
Molti bambini grandi-prematuri (< 25 settimane ) presentano disfunzioni neurologiche e anomalie di sviluppo. La prematurità (soprattutto nei soggetti con peso al a nascita infe- riore a 1500 g) è una condizione di elevato rischio per la sordità, così come per altri disor-dini: ciò comporta che fra questi "bambini fragili" siano frequenti i casi con handicap multi- pli. Studi longitudinali che hanno considerato bambini prematuri e di peso molto basso hanno evidenziato che a 5 anni questi presentavano un tasso di ipoacusia 5 volte maggio- re rispetto ai bambini della stessa età ma senza problemi al a nascita. Nonostante il mi- glioramento dei trattamenti medici rivolti ai neonati, si ritiene ancora che un peso al a na- scita inferiore a 1500 g, una età gestazionale inferiore a 31 settimane, l'essere un neona- to sottoposto a terapia intensiva, siano altrettanti indicatori altamente predittivi per la ipoa- cusia infantile(51). b) CMV e altre infezioni virali
Il citomegalovirus (CMV) nel a sua forma sintomatica e asintomatica, è la più fre- quente causa di infezione congenita. Nei paesi sviluppati il CMV è a tutt'oggi il solo impor- tante agente virale che può causare la sordità, poiché le vaccinazioni contro rosolia, paro- tite, morbil o hanno praticamente fatto scomparire i casi sostenuti dai rispettivi virus. La sordità causata da CMV può mostrare un esordio ritardato, fino a 6 anni di età, e un dete- rioramento progressivo del a funzione uditiva. Per questi casi è necessario programmare una sorveglianza sufficientemente prolungata. È da rilevare che spesso (70%) nel e for- me sintomatiche sono documentabili lesioni del sistema nervoso centrale, responsabili di sequele neurologiche che possono accompagnarsi al a sordità. Una ricerca condotta in Italia(52-53) dimostra che il 10% dei bambini di età inferiore a 2 mesi, con ipoacusia neurosensoriale, ha un'infezione congenita da citomegalovirus. La diagnosi era ottenuta attraverso la ricerca del DNA virale con il dosaggio del a PCR, u- sando i campioni di sangue utilizzati per il test di Guthrie. Negli USA un'indagine condotta su neonati che non superano i test di screening per sordità congenita e hanno una coltura positiva nel e urine per CMV, evidenziata con anticorpi monoclonali(54), metteva in eviden- za che il 6% era affetto da infezione congenita. Nel primo rapporto completo sul 'eziologia del a sordità congenita, in un campione molto rilevante di neonati, ben 87.000 di cui 170 inviati al 3° livel o(20), risultava che è pos- sibile ottenere una diagnosi eziologica nel 55,2% dei soggetti e fra questi il 18,8% era af- fetto da infezione congenita da CMV, il 10,3% del totale (Figura 3). Se è vero che l'infezione da CMV è la più frequente causa di infezione congenita nel neonato, l'aumento dei casi appare probabilmente legato al sempre più frequente utilizzo di test diagnostici molto accurati(55). Una diagnosi precoce nel e forme sintomatiche assume una particolare importanza al fine di intraprendere in modo sol ecito un trattamento antivira- le con ganciclovir/vanganciclovir(56). Lanari et al.(57) hanno dimostrato che alti livel i di viremia nel periodo neonatale, in soggetti le cui madri avevano avuto durante la gravidanza un'infezione primaria, ricorrente o non definita da CMV, costituivano un fattore altamente predittivo per la presenza di se- quele a distanza e quindi anche in grado di condizionare scelte terapeutiche nel neonato sintomatico e asintomatico. Il danno uditivo nei soggetti con infezione congenita o inapparente da CMV può ri- manere stabile, fluttuare, progredire in più del 50% dei casi o talvolta svilupparsi più tardi nell'infanzia; un regolare fol ow-up audiometrico è pertanto raccomandato fino a 6 anni, ma una particolare attenzione va garantita fino al 'età adolescenziale; in caso di ipoacusia grave-profonda trova una indicazione precisa l'impianto cocleare (58). È comunque eviden- te che solo una prevenzione primaria del 'infezione materna con un efficace vaccino po- trebbe risolvere un problema di difficile soluzione quale quel o dell'infezione da CMV, im- portante causa di sordità e di danno neuromotorio nel neonato. Molto incoraggianti ap- paiono i risultati ottenuti con un recente vaccino (legato al a glicoproteina BI del CMV), somministrato a donne sieronegative un anno prima del a nascita del bambino, che si è di- mostrato capace di diminuire il numero dei casi di infezione materna e congenita da CMV, con una efficacia pari al 50% (59), ma siamo ancora molto lontani da un'applicazione pratica. c) Farmaci ototossici
È ben conosciuto il ruolo di certe categorie di farmaci (ad esempio antibiotici amino- glicosidici) nel causare danni cocleari irreversibili e quindi una sordità. In studi recenti tut- tavia il nesso causale tra questi farmaci e la sordità infantile non appare così stretto come si pensava in passato. Almeno fra i neonati grandi prematuri, sembra che la coesistenza di altri fattori di rischio per la sordità sia più importante nel determinare il danno, piuttosto che il fattore "farmaco" isolatamente considerato. Individui portatori di alcune mutazioni mitocondriali, sottoposti a terapia con aminoglicosidici, possono andare incontro ad ipoa- cusia bilaterale, da severa a profonda, anche dopo una singola dose(40). 3.4 Meningite
Se la meningite batterica, come causa di sordità infantile, è attualmente diventata sempre meno frequente (1-2%) nei paesi a sviluppo avanzato, grazie al e campagne vac- cinali, è ampiamente documentato che una del e più frequenti e devastanti sequele del e meningiti batteriche è certamente il danno uditivo, esso si riscontra tra il 7% e il 35 % dei sopravvissuti. L'interessamento appare molto elevato, dal 30% al 50%, nel e meningiti pneumococciche, tra il 10% e il 30% nel e forme da haemophilus influenzae di tipo B, e tra il 5% e il 25% nel e meningiti meningococciche(60-61). La sordità secondaria a meningite è causata da una diffusione del 'infezione dal labi- rinto alle meningi attraverso l'acquedotto cocleare, o per danno diretto del nervo cocleare. L'ipoacusia da meningite è nel a maggior parte dei casi bilaterale, frequentemente di gra- do severo o profonda. Nei bambini può porre notevoli problemi riabilitativi, data la possibi- lità di lesioni al nervo cocleare che impediscono di sfruttare appieno l'amplificazione forni- ta da una protesi acustica. Indicatori prognostici di sordità sono considerati uno stato di coma e la presenza di alterazioni a livel o del labirinto rilevate alla TAC e al a Risonanza magnetica(62). Una te- mibile complicazione del a meningite è infatti costituita dall'ossificazione del labirinto, che sembrerebbe sempre iniziare a livel o del canale semicircolare laterale; quando ciò si veri- fica è indicato un impianto cocleare da eseguire precocemente, anticipando la completa obliterazione ossea del dotto cocleare. Uno studio audiometrico (con le modalità adatte al 'età del paziente, solitamente mol- to piccolo) deve essere eseguito a tutti i pazienti che hanno avuto una meningite batterica, che comunque devono essere attentamente control ati nel tempo. Un problema particolare è rappresentato dal 'insorgenza di una meningite batterica, spesso associata ad una otite media acuta, nei soggetti sottoposti ad impianto cocleare, soprattutto se di età inferiore a tre anni e nei primi due mesi dopo il trapianto. Per quanto si sia riscontrato che questo può avvenire prevalentemente in casi di malformazioni co- cleari (ed in precedenza con un particolare impianto dotato di "positioner" che non è più in commercio), i soggetti con sordità profonda, candidati al 'impianto, devono essere sotto- posti ad una adeguata vaccinazione antipneumococcica ed anti-haemophilus influenzae tipo B, oltre alla vaccinazione contro l'influenza e la meningite da meningococco(63-65).
4. LO SCREENING AUDIOLOGICO NEONATALE
4.1 In passato la sordità nei primi giorni di vita veniva ricercata con tecniche di audio-
metria comportamentale. La valutazione veniva effettuata prevalentemente sui neonati a
rischio e permetteva di individuare solo le sordità bilaterali profonde. Questa tecnica è sta- ta attualmente abbandonata per la scarsa sensibilità e specificità del test.
4.2 Le nuove tecniche di screening
4.2.1 Le emissioni otoacustiche (TEOAE, DPOAE)
Quando un suono raggiunge un orecchio normale si verifica un'eccitazione di alcune specifiche cel ule presenti nel a coclea che si contraggono ed emettono un "rumore" di ri- torno che può essere registrato. Su tale base è stata elaborata una metodica che va sotto il nome di "emissioni otoacustiche": nel condotto uditivo esterno si inserisce un sondino at- traverso il quale si invia un suono che giunge al a coclea; lo stesso sondino è in grado di registrare il segnale di ritorno emesso dal e cel ule cocleari. La mancanza di tale segnale implica un'anomalia del a funzione di tali cel ule, che rappresenta, quasi sempre, il disturbo al a base del a sordità neurosensoriale infantile. L'avvenuta registrazione del segnale di ri- torno è invece la dimostrazione che il soggetto sottoposto al 'indagine ha una normale fun- zionalità del a coclea che, nei soggetti senza fattori di rischio, si correla solitamente a una normale capacità uditiva. La rapidità di esecuzione, l'assenza di fastidio e l'affidabilità ren- dono questo test uno strumento valido per lo screening delle ipoacusie in età neonatale. Nel caso in cui le emissioni otoacustiche siano presenti (pass), è possibile affermare che la coclea funziona correttamente e, in genere, non è necessario eseguire altri esami. Vice- versa se le emissioni otoacustiche risultassero assenti (il risultato negativo potrebbe di- pendere dal e difficoltà di registrazione, dal e caratteristiche anatomiche particolari del ne- onato, dal a presenza di cerume, da un'infiammazione del 'orecchio o da una reale soffe-renza della coclea), è necessario ricorrere ai potenziali evocati uditivi. L'esecuzione del e TEAOE richiede pochi minuti e si esegue preferibilmente durante il sonno del neonato. L'addestramento del personale che eseguirà il test richiede solita- mente poche ore, occorre però un periodo di training per migliorare le performance. Oggi sono comunemente impiegate delle apparecchiature automatiche per otoemissioni che possono essere utilizzate anche da personale non altamente specializzato, il risultato del test comparirà sul display. Le apparecchiature utilizzate permettono di avere una docu- mentazione cartacea del risultato del test; sono inoltre interfacciabili con un personal com- puter tramite una porta USB, così che i dati possano essere archiviati da ogni punto- Se non vengono registrate otoemissioni (refer) il test va ripetuto nei giorni successivi, tenuto conto del o schema organizzativo adottato e del e apparecchiature disponibili. L'American Academy of Pediatrics fissa uno standard per i soggetti da ricontrollare (refer) < al 4% dei neonati sottoposti al o screening, alcuni ospedali utilizzano questa misura per monitorare la qualità del programma di screening. Si tratta, come si vede, di un test non invasivo semplice e che permette di registrare le emissioni evocate prodotte naturalmente dalla coclea e dopo stimolazione. La sensibilità è quasi del 100%, la specificità è molto e- levata: 97% (solo tre falsi positivi)(66-67). Numerose società scientifiche, come l'American Academy of Pediatrics, il National Institutes of Health e l'European Consensus Development Conference on Neonatal Hea-ring Screening, ne hanno raccomandato l'impiego come metodica di screening universale, anche tenuto conto che sono completamente soddisfatti i criteri per giustificare uno screening universale. Perché un programma di screening uditivo neonatale sia efficace occorre comunque rispettare le seguenti condizioni: - Poter esaminare almeno il 95% della popolazione dei neonati che si intendono screena- - I soggetti refer al termine del o screening non devono essere più del 4% - I falsi positivi non devono essere più del 3% - I falsi negativi devono essere uguali a 0 - La percentuale dei soggetti persi al fol ow-up non deve essere superiore al 5%. Lo screening uditivo universale è già stato adottato da numerosi paesi in Europa, quel i che hanno raggiunto e superato il 90% dei neonati nel 2006 sono: Austria, Belgio (Fiandre), Croazia, Inghilterra, Lussemburgo, Olanda, Polonia e Svezia; negli USA più del 90% dei neonati viene sottoposto a screening.
4.2.2 I potenziali evocati uditivi del tronco (ABR)
Si tratta di una metodica sicuramente più complessa, cui è necessario ricorrere nel caso in cui non è stato superato lo screening. L'ABR (Auditory Brainstem Response), in epoca neonatale o comunque nei primi mesi di vita, rappresenta la prima tappa diagnosti- ca per i soggetti che non hanno superato lo screening nei casi di soglia uditiva aumentata. Quando un suono raggiunge l'orecchio, attiva prima le strutture periferiche (orecchio medio e le già ricordate strutture cocleari del 'orecchio interno) e poi le strutture retroco- cleari (nervo acustico e tronco encefalico): a tale attivazione corrisponde la formazione di un segnale elettrico che può essere registrato. Per eseguire l'ABR, al bambino vengono applicati quattro piccoli elettrodi adesivi in punti stabiliti del cranio; quindi, attraverso una normale cuffia, si invia un suono di variabile intensità. Come si accennava, le strutture presenti lungo il decorso del e vie uditive generano delle onde (le più importanti sono cin- que) che vengono registrate da un computer. Dal 'analisi di tali onde è possibile stabilire se il piccolo ha un udito normale o meno. In caso di ipoacusia i potenziali evocati uditivi del tronco consentono di risalire al a soglia uditiva con notevole precisione. Nel a pratica clinica vengono distinte due modalità di registrazione dei potenziali evo- cati uditivi troncoencefalici: l'ABR e l'A-ABR o Automated Auditory Brainstem Response. Mentre la prima metodica consente una precisa determinazione del a soglia uditiva e una diagnosi definitiva di ipoacusia, la seconda è utilizzata per alcune categorie di pazienti come procedura di screening in associazione al e Otoemissioni (v. più avanti). La regi- strazione automatica dei potenziali uditivi evocati da uno stimolo acustico viene attuata in pochi minuti mediante uno strumento portatile, ma non presenta le caratteristiche di affi- dabilità e specificità necessarie perché sia utilizzata nella diagnosi di soglia uditiva. L'ABR, che permette di rilevare i livel i di attività elettrofisiologica generati in risposta ad uno stimolo sonoro, non è invasivo, deve essere condotto mentre il bambino dorme, ri-chiede un tempo maggiore di analisi rispetto ai test di screening come TEOAE o A-ABR e può essere eseguito solo da personale specializzato e sotto la supervisione di uno specia- lista audiologo. L'ABR rappresenta l'indagine di secondo livel o riservata ai soggetti che non passano lo screening (refer), in considerazione del 'elevato costo del e attrezzature e del a necessi- tà di personale specializzato per l'esecuzione del test. Nei bambini ricoverati nelle Unità di terapia intensiva neonatale (NICU) l'AABR (ABR automatico) è consigliato come test di screening in quanto più sensibile a riconoscere an- che le perdite uditive retro-cocleari, assai frequenti in questi soggetti, ma molto rare nei bambini sani a termine. Il test comporta un maggior costo di materiale, richiede uno stato di veglia quieta o di sonno nel bambino e più tempo per l'esecuzione.
4.2.3 Ulteriori approfondimenti diagnostici
In presenza di patologie del sistema nervoso centrale, l'ABR non permette di giunge- re a una diagnosi definitiva né di raccogliere le informazioni utili ai fini di una corretta pro- tesizzazione acustica. Nei casi selezionati si utilizzano metodiche più avanzate, come le Auditory Steady-State Response (ASSR) e l'elettrococleografia, al fine di chiarire meglio la configurazione di soglia e la reale entità del a perdita uditiva. Questi esami devono essere eseguiti presso i Centri di Riferimento individuati dal a Programmazione Regionale. Auditory steady-state response Si tratta di potenziali evocati uditivi che utilizzano stimoli specifici in frequenza e che forniscono una informazione dettagliata del a soglia uditiva. Ciò risulta particolarmente uti-le per la corretta applicazione del 'ausilio protesico. Elettococleografia È la metodica elettrofisiologica più complessa che consente una diagnostica assolu- tamente precisa sia per ciò che concerne l'entità del a perdita sia la sede della lesione. Va praticata in anestesia generale quando sono assenti le risposte ai potenziali evocati tron- coencefalici o, comunque, laddove tali risposte non siano sufficienti. 5. LA SITUAZIONE ITALIANA
In Italia la diagnosi di sordità infantile viene effettuata ancora con notevole ritardo. Un'indagine epidemiologica condotta nel 2003-2004(68) dimostrava che in Italia solo il 29,3% dei neonati veniva sottoposto a uno screening neonatale. Un aggiornamento dei dati relativi al 'anno 2006(69) mette in evidenza che il numero dei soggetti esaminati al a nascita era pari al 48,4% (Nord-Ovest 79,5%, Nord-Est 57,2%, Centro 32,0%, Sud 42,6%, Isole 11,3%). Il sempre maggiore impegno dei pediatri- neonatologi italiani in questo settore fa ritenere che nel 2008 lo screening del a sordità congenita venga effettuato a circa il 60% dei neonati. Solo 5 regioni italiane hanno delibe- rato, con norme attuative, lo screening uditivo neonatale: le prime sono state la Liguria e la Campania, a cui si sono recentemente affiancate la Basilicata (con molti problemi irri- solti), le Marche e la Toscana. Altre regioni come la Lombardia, il Veneto, la Provincia A. di Bolzano, l'Emilia-Romagna, il Friuli-Venezia Giulia e l'Umbria hanno iniziato ad effettua- re un'analisi del a situazione esistente ed alcune anche ad elaborare documenti tecnici di indirizzo, al fine di rendere operativo lo screening neonatale del a sordità congenita, defi- nendo anche il percorso del paziente affetto da sordità congenita o acquisita. Colpisce la mancanza di iniziative regionali in Piemonte, nella Provincia A. del Trentino-Alto Adige, in Lazio, Molise, Calabria, Sicilia, Puglia e Sardegna, anche se molti reparti o sezioni di ne- onatologia effettuano lo screening, sia pure in modo incompleto. Vanno segnalate alcune esperienze-pilota, come ad esempio quel a condotta dal a clinica Mangiagal i di Milano, in col aborazione con l'Istituto di ingegneria biomedica del CNR(70), nel 'ambito di un progetto di ricerca (Progetto Milano). 6. IL PROTOCOLLO DEL PROGRAMMA DI SCREENING NEONATALE
UNIVERSALE
Il programma di screening uditivo neonatale universale prevede una strutturazione in tre livel i assistenziali, ciascuno eseguito in centri adeguatamente attrezzati. Il primo livel o assistenziale è rappresentato da tutti punti nascita pubblici e privati accreditati Il secondo livel o è rappresentato dai Servizi di Audiologia e Foniatria, autonomi o aggre- gati al e U.O. di Otorinolaringoiatria del S.S.N. Il terzo livel o assistenziale è rappresentato dal Centro o dai Centri di riferimento regionali. Esistono percorsi diagnostici diversi, a seconda che si tratti di neonati sani ("wel babies") o bambini ricoverati in Terapia Intensiva Neonatale (TIN).
6.1 Il protocollo di screening per neonati sani (well babies)
In condizioni di sonno spontaneo, oppure durante la poppata, nel 'ambiente più si- lenzioso possibile, in 2°-3° giornata dal a nascita vengono effettuate le otoemissioni acu- stiche evocate (TEOAE). Se le otoemissioni sono presenti da entrambi gli orecchi, il neo-nato viene definito "pass" al test e la procedura di screening ha termine (a parte i casi che rientrano nelle categorie a rischio audiologico, vedi dopo). Nel caso in cui le otoemissioni siano assenti in uno o entrambi gli orecchi, la risposta viene definita "refer" ed il test viene ripetuto entro le tre settimane. In questa occasione, sarebbe ottimale ripetere il test con le otoemissioni insieme ai potenziali uditivi evocati (ABR) da screening (ABR automatico o A- ABR, two-step protocol), eseguibili spesso con la medesima apparecchiatura; ove ciò non fosse possibile, si ripeteranno le sole TEOAE. Se le TEAOE sono presenti e risultano pass in entrambi gli orecchi, la procedura è da ritenersi conclusa (a parte i casi che rientrano nel e categorie a rischio audiologico, vedi dopo). Nel caso di conferma di risposta "refer" si invieranno i pazienti al centro di secondo livel o per gli accertamenti audiologici consistenti nel 'esecuzione degli AABR/ABR per so- glia che devono essere eseguiti entro il secondo/terzo mese di vita. I piccoli pazienti risultati refer al a procedura di screening di secondo livel o (otoemissioni ed AABR/ABR) vengono inviati ed esaminati entro il terzo/quarto mese di vita nel centro di terzo livello per la diagnostica audiologica completa in termini di grado di perdita, sede di lesione, eziopatogenesi e disabilità comunicative. 6.2 Il protocollo di screening per i neonati ricoverati in TIN
Il test di screening consigliato per tutti i neonati ricoverati in un reparto di terapia in- tensiva/patologia neonatale (TIN) prevede la contemporanea esecuzione delle TEAOE e degli A-ABR, in quanto questi sono a maggior rischio di sviluppare una neuropatia uditi- va(71), situazione nel a quale il danno è situato a livel o del e cel ule ciliate interne o del e vie nervose e nel a quale, pertanto, il test con le sole otoemissioni potrebbe risultare nor- male (le TEAOE infatti valutano la funzionalità del e sole cel ule ciliate esterne della co- clea). Nel caso di risposta refer è consigliabile eseguire almeno due volte il test prima del a dimissione. I neonati che risultano refer ad uno o entrambi gli orecchi, verranno inviati, en- tro il terzo/quarto mese di vita corretto per l'età gestazionale, presso un centro di riferimen- to di terzo livel o per l'approfondimento diagnostico. I neonati che risultino invece pass, ma che rientrano nel e categorie a rischio (v. tab. 1), verranno sottoposti a periodici control i uditivi semestrali nel corso dei primi 5 anni di vita. Tabella 1: Fattori di rischio per ipoacusia progressiva o ad esordio tardivo presenti alla 1. Storia familiare* positiva per ipoacusia infantile permanente. 2. Ricovero in NICU per un periodo superiore a 5 giorni o ciascuna delle se- guenti condizioni, indipendentemente dal a durata del ricovero in NICU: EC- MO*, ventilazione assistita, assunzione di farmaci ototossici (gentamicina e tobramicina) o diuretici del 'ansa (furosemide/Lasix), iperbilirubinemia che ha reso necessaria l'exanguinotrasfusione. 3. Infezioni intrauterine, quali CMV*, herpes, rosolia, sifilide e toxoplasmosi. 4. Malformazioni craniofacciali, incluse quel e del padiglione auricolare, del condotto uditivo esterno, appendici e fistole pre-auricolari e anomalie dell'osso temporale. 5. Anomalie quali ciuffo di capel i bianchi, che sono descritte in associazione con sindromi che includono ipoacusia permanente neurosensoriale o tra- 6. Sindromi associate con ipoacusia progressive o ad esordio tardivo*, come neurofibromatosi, osteopetrosi, sindrome di Usher; altre sindromi frequente- mente identificate includono la sindrome di Waardenburg, Alport, Pendred, Jervel e Lange-Nielsen. 7. Disordini neurodegenerativi*, quali la sindrome di Hunter, o neuropatie sensi- tive-motorie, come la atassia di Friedreich e la sindrome di Charcot-Marie-
7. LA SORVEGLIANZA NELLE ETÀ SUCCESSIVE: IL PROBLEMA DEL-
LE FORME AD INSORGENZA TARDIVA O PROGRESSIVA E DELLE
CATEGORIE A RISCHIO
Ai test di screening neonatali attualmente utilizzati possono sfuggire i bambini che presentano forme di ipoacusia progressiva o ad insorgenza tardiva e le neuropatie uditive. Non sono disponibili dati Italiani sul 'incidenza di queste forme di ipoacusia, ma stime e- seguite in altri Paesi suggeriscono che la percentuale di ipoacusie che si sviluppano dopo il periodo neonatale, e che non possono pertanto essere identificate con il test di screening, possa rappresentare fino al 20-25% del totale(72). Come consigliato dal JCIH 2007(73), alcune categorie di pazienti che presentano più alto rischio di sviluppare una ipoacusia devono essere sorvegliati con particolare attenzio- ne dai pediatri di famiglia e/o nei centri audiologici. Pertanto i bambini che presentano fat- tori di rischio per ipoacusia progressiva o ad esordio tardivo indicati nel a tabel a 1 devono essere sottoposti ad un monitoraggio. In particolare, quei bambini che presentano fra que- sti fattori di rischio quel i indicati con l'asterisco devono essere sottoposti ad una valutazio-ne nel centro audiologico ogni 6 mesi-1 anno, essendo particolarmente elevato il rischio di sviluppare ipoacusia. Il responsabile del o screening del punto nascita o del a NICU deve segnalare al momento del a dimissione al pediatra di famiglia l'appartenenza del bambino ad una di queste categorie a rischio. È inoltre consigliabile che i bambini che hanno sviluppato una del e condizioni riportate nella tabel a 2 siano inviati presso un centro audiologico di 2° livel o per la valutazione. Tabella 2: Fattori di rischio per ipoacusia ad esordio tardivo che possono presentarsi nel periodo post-natale. 1. Infezioni post-natali associate con ipoacusia neurosensoriale, incluse la me- ningite batterica e virale (soprattutto da herpes virus e varicel a). 2. Traumi cranici, soprattutto fratture a carico del basicranio e del 'osso tempo- rale che richiedono ricovero in ospedale. 3. Otite media essudativa (OME) ricorrente o persistente per più di 3 mesi. 4. Terapia con farmaci ototossici (soprattutto chemioterapici, amino glicosidici). 5. Preoccupazione degli educatori riguardo l'udito, la percezione verbale, lo svi- luppo del linguaggio o ritardi di sviluppo. Se le forme postnatali rappresentano il 20-25% del e sordità bilaterali del bambino, in alcuni di questi soggetti (26%) non si evidenziavano fattori di rischio, in altri il peggio- ramento della funzione uditiva si verificava dopo i tre anni. Il monitoraggio dei soli bambini a rischio non è pertanto sufficiente; quindi, indipendentemente dal risultato negativo del o screening audiologico neonatale, tutti i programmi per la tutela del a salute infantile, nei primi anni di vita, devono prevedere un attento control o del 'acquisizione del linguaggio nel bambino, evidenziando precocemente una possibile diminuzione del 'udito. I genitori sono la prima linea di sorveglianza, per questa ragione deve essere utilizzata ogni oppor- tunità per fornire loro informazioni chiare e comprensibili sul 'acquisizione delle varie tappe del linguaggio e sul a funzionalità del 'apparato uditivo nel bambino, anche attraverso la somministrazione di un apposito questionario sul 'udito, in funzione del e varie età del bambino, in occasione dei control i di salute. I pediatri di famiglia, gli operatori sanitari di area pediatrica e gli insegnanti devono pertanto essere fortemente impegnati nei pro- grammi di sorveglianza audiologica. Durante i bilanci di salute deve essere sempre ricer-cato un eventuale ritardo nel e tappe di sviluppo dell'apparato uditivo e del linguaggio, ri- cordando che l'esecuzione di un test di distrazione come il Boel Test, prevista dal 'accordo col ettivo nazionale dei pediatri di famiglia, costituisce un test generale di monitoraggio del- lo sviluppo psicomotorio, ma ha una troppo bassa sensibilità e specificità come test uditivo al '8° mese di vita ed è troppo tardivo per una diagnosi di sordità. Si raccomanda di effettuare le indagini audiologiche anche ogni volta che si pone un sospetto di Ritardo semplice del linguaggio o di Disturbo Specifico del Linguaggio o dell'Apprendimento, ogni volta che un bambino non raggiunge - prime parole entro i 2 anni - frase bitermine entro i tre anni - intel igibilità del a produzione verbale > 75% entro i 4 anni. Le tappe del o sviluppo uditivo e del linguaggio sono riportate nel a Tabel a 3. Tabella 3: Le tappe dello sviluppo uditivo e del linguaggio Dal a nascita a 3 mesi Sobbalza in caso di suoni forti Si sveglia ai suoni (rumori) Ammicca o spalanca gli occhi in risposta ai suoni (riflesso) Si calma sentendo la voce del a mamma Smette di giocare sentendo nuovi suoni Cerca la fonte di nuovi suoni che sono fuori dal a sua visuale Si diverte con giochi musicali Emette suoni con inflessione Dice "mama" Risponde al suo nome Sa riconoscere il "no" Esegue ordini semplici Usa in modo espressivo un vocabolario di 3-5 parole Imita alcuni suoni Sa indicare le parti del corpo Usa in modo espressivo frasi di 2 parole (con vocabolario di 20-50 parole) Il 50% del linguaggio è comprensibile da un estraneo Usa in modo espressivo frasi di 4-5 parole (con vocabolario di 500 parole) L'80% del linguaggio è comprensibile da un estraneo Comprende il significato di alcuni verbi Pediatrics 2003(51), modificato La check list può essere utilizzata dai pediatri, dagli operatori sanitari di area pediatrica, dagli insegnanti e nel a elaborazione dei questionari da sottoporre ai genitori al fine di individuare i soggetti a rischio da inviare ad una valutazione audiologica. 8. IL PERCORSO DEI BAMBINI CON IPOACUSIA NEUROSENSORIALE(*)
Il percorso dei pazienti con ipoacusia neurosensoriale si presenta ovviamente diffe- renziato a seconda se l'ipoacusia neurosensoriale sia mono o bilaterale. Le forme monola- terali, dato che non determinano particolari alterazioni né del o sviluppo del linguaggio né di quello psicomotorio e cognitivo, sono monitorate con successivi fol ow-up, per poter in- tervenire rapidamente qualora dovessero evolvere in forme bilaterali. I control i sono an- nuali fino ai 6 anni. Per le forme bilaterali, il protocollo prevede che tutte le ipoacusie superiori ai 60 dB vengano sottoposte ad immediata terapia protesica, mentre per le forme comprese tra i 50 ed i 60 dB è previsto un retest a distanza di un mese per un'ulteriore conferma del livel o uditivo. Ciò si è reso necessario in quanto, soprattutto nei bambini fortemente pretermine, si evidenziano lievi oscil azioni nel a soglia uditiva che, in alcuni casi, possono inficiare la precisione diagnostica. Per quel che concerne, invece, i pazienti con soglia inferiore ai 50 dB, il protocol o prevede che questi debbano essere seguiti nel tempo con fol ow-up periodici (inizialmente trimestrali e successivamente semestrali) da eseguirsi fino al a determinazione di una so- glia audiometrica "affidabile". Il monitoraggio deve prevedere, quando possibile, la determinazione della soglia udi- tiva in tutte le frequenze del campo uditivo per la via aerea e per la via ossea con accurata valutazione del gap trasmissivo in relazione ai risultati del a timpanometria. 8.1 La presa in carico nel percorso riabilitativo
Un team multidisciplinare composto da audiologo-foniatra, audiometrista, logopedista e audioprotesista, psicologo, pediatria e neuropsichiatra infantile, prende in carico da subi- to il bambino protesizzato e la famiglia, al fine di fornire ulteriori spiegazioni sul a funziona-lità del 'ausilio protesico e sul percorso riabilitativo. La presa in carico si svolge in occasio-(*) I Capitoli 8-9-10-11-12 fanno più strettamente riferimento a quanto elaborato dal Gruppo di Lavoro nominato dal Dipartimento Programmazione del Ministero della salute nel 2010, resp. Dr. Alessandro Ghirardini, costituito da: Alessandro Martini (coordinatore), Stefano Berrettini, Sandro Burdo, Alessandro Ghirardini, Paola Marchisio, Elio Marciano, Lodovico Perletti, Nicola Quaranta. ne del primo control o protesico, ma il team continua a lavorare con il bambino e la fami- glia in genere fino al compimento del primo anno di età. Il lavoro svolto riguarda la valuta- zione del e competenze acustico-percettive, comunicative e cognitivo-linguistiche del bambino e la stesura di un programma di intervento, che viene proposto e condiviso con i col eghi del a riabilitazione delle ASL, ai quali i bambini devono essere inviati dopo il per- corso iniziale presso il Centro Regionale di Riferimento. Il nucleo familiare va coinvolto in modo attivo, deve ricevere informazioni su come stimolare il bambino per attivare, al enare e/o potenziare le competenze summenzionate, in armonia con le tappe fisiologiche del o sviluppo, e viene responsabilizzato in ogni tappa del percorso riabilitativo.
9. ASPETTI GESTIONALI E ORGANIZZATIVI
La semplicità del e tecniche diagnostiche di base (TEAOE) per l'esecuzione del o screening del a sordità congenita permette una diagnosi molto precoce della sordità neo- natale; tuttavia vale la pena di esaminare più in dettaglio il model o organizzativo(74) che, partendo dal risultato di un test (refer) neonatale deve accompagnare tutto il percorso del paziente affetto da sordità congenita (Figura 3). 9.1 Il Punto nascita (I livello)
La prima fase è certamente la formazione del personale infermieristico del Nido. Non vengono richiesti alti livel i di specializzazione, possono essere sufficienti 5-6 ore di for- mazione, occorre tuttavia un periodo più lungo di addestramento per raggiungere i livel i di qualità previsti per lo screening. È indispensabile il col egamento con il Centro di II livello per la conferma del a dia- gnosi e con il Centro regionale di riferimento per la sordità congenita (III livel o). Al 'interno del a struttura neonatale vanno definite le figure professionali incaricate di eseguire lo screening. Deve essere indicato il medico responsabile del o screening che dovrà stabilire le procedure, il fol ow-up e l'organizzazione dei flussi informativi. Deve es- sere stabilito il consenso-control o per un secondo esame ai refer, entro 3 settimane dal a nascita. Va ricordato che è indispensabile una informazione adeguata dei genitori circa il significato dello screening e le finalità del o stesso che deve comprendere il consenso in- formato anche con una modulistica adeguata che deve prevedere la comunicazione dei risultati. 9.2 La conferma della diagnosi (II livello)
A questa struttura (Servizi o Centri di audiologia di II livel o) devono essere inviati i soggetti che non passano lo screening (refer) per l'esecuzione degli ABR soglia. Questa tecnica può essere eseguita solo da personale specializzato e con la supervisione di uno specialista audiologo. Le terapie intensive neonatali devono poter disporre di questo per- sonale che è, d'altra parte, presente nel e Aziende Ospedaliere che dispongono di un Servizio di audiologia. Anche in questo caso deve essere identificata una figura profes- sionale medica che stabilisca con la Direzione Sanitaria le procedure, gli standard del e apparecchiature e la disponibilità del personale. Deve sempre essere prevista una col aborazione permanente, anche per via infor- matica, con i punti nascita afferenti ed il Centro regionale di riferimento. 9.3 I Centri di riferimento regionali (III livello)
Il Centro o i Centri di audiologia di riferimento regionali (al 'incirca 1 ogni 2-3 milioni di abitanti) devono non solo assicurare il necessario approfondimento diagnostico, ma ga- rantire anche la formazione del personale dei punti nascita e dei Centri di II livel o, nonché l'organizzazione e il monitoraggio di tutto il processo di screening. I Centri di riferimento regionali, in col aborazione con i centri di II livel o, il personale dei servizi territoriali ed i pediatri di famiglia, devono inoltre prevedere: - Regolari control i del o sviluppo del a percezione uditiva e del a abilità comunicativa e linguistica e psicomotoria dei pazienti. - Linee guida per la protesizzazione e la corretta indicazione all'impianto cocleare. - Sostegno psicologico e sociale alla famiglia. - Graduale informatizzazione di tutti i flussi informativi al fine di mettere in rete, in tempo reale, famiglie, strutture sanitarie, pediatri di famiglia e tutti gli operatori sanitari coinvolti nel a diagnosi e cura del a sordità infantile, ricorrendo anche al a telemedicina. Le figure professionali che devono far parte del 'equipe di un Centro di riferimento regionale, oltre ai responsabili del Centro, sono costituite da: - medici audiofoniatri - Unità di segreteria - informatici Il loro numero deve essere correlato con i volumi di attività. Deve inoltre essere pre- visto uno stretto col egamento con i Servizi di genetica clinica, sindromologia, pediatria e infettivologia. Oggi nessun centro di Audiologia e Otologia può prescindere dal a col abo-razione con il genetista clinico e con la diagnosi di genetica molecolare(75); questa col abo- razione sarà probabilmente ancor più necessaria nel prossimo futuro sia per un miglior in- quadramento diagnostico, sia per un miglior trattamento riabilitativo, farmacologico (tera- pia genetica, cel ule staminali, ecc.). 10. I COSTI DELLO SCREENING NEONATALE DELLA SORDITÀ
10.1 Punto nascita
L'analisi dei costi è relativa ad un model o organizzativo di screening universale, da condurre nei punti nascita, che preveda la registrazione del e otoemissioni, con previsione di un retest nel 4% dei casi entro la dimissione o nelle prime 2-3 settimane di vita, a se- conda dei model i organizzativi adottati e dopo che il personale infermieristico abbia supe- rato la fase di addestramento. A) Il costo medio di uno strumento per screening con otoemissioni è di 5.000 €, IVA compresa (prezzi fra circa 3.800 e 4.600 €). B) Costo di istruzione personale (esecutori dello screening e gestore dei dati): 5-6 ore, TOTALE COSTI INIZIALI (A+B) = 6.000 € Ammortamento in 5 anni: 1.200 € al 'anno
- Costi vivi
I costi vivi comprendono le spese per il personale, i materiali, la manutenzione e la gestione dei dati. A) Il tempo medio per la registrazione del e otoemissioni automatiche da entrambe le o- recchie in un neonato è di 4'-5'. Occorre tenere conto poi del e manovre di preparazio- ne e di alcuni tempi morti per aspettare le condizioni ambientali acustiche più favorevo- li, inoltre il 4% dei casi deve eseguire un retest. Il tempo medio richiesto può essere stimato in circa 10'-12'. Considerato un costo orario lordo per infermiere professionale di circa 13 euro, il costo per bambino è di 2,5 euro. B) Costo di materiale di consumo (sonde): 0,5 euro/caso C) a - Costi del personale responsabile dei dati (immissione del dato, richiamo dei "refer" ed invio per ABR con appuntamento). Stimando un tempo medio di 5' per caso ed un costo orario lordo di 13 euro, si può stimare un costi di 1 euro per caso. b - Costi di cancelleria-telefono: circa 1 euro a caso. Totale = 2,0 €/bambino TOTALE COSTI VIVI (A+B+C) = 2,5 + 0,5 + 2,0 = 5,0 €/bambino Esempio di costo annuale per un punto nascita con 1.000 neonati all'anno
Costo fisso con ammortamento in 5 anni Costo vivo per 1.000 neonati Costo unitario per bambino = 6,2 euro Se è possibile, come abbiamo visto, fare in linea teorica una stima dei costi del o screening, in realtà il costo reale dipende non solo dalla qualifica del personale impiegato, dal tipo di apparecchiature utilizzate e dal e caratteristiche del punto nascita (numero dei nati, presenza di NICU ecc.), ma anche dai risultati effettivamente ottenuti, come il numero dei neonati che non hanno superato lo screening (refer), il numero dei soggetti persi al fol- low-up e la percentuale del a popolazione sottoposta al o screening. 10.2 La conferma della diagnosi
I soggetti che non hanno superato lo screening (refer) devono essere inviati a un se- condo livel o per l'esecuzione degli ABR soglia. Questa tecnica può essere eseguita solo da personale specializzato e con la supervisione di uno specialista audiologo. Questa pro- cedura deve essere effettuata entro i 3 mesi di età; stimando che i soggetti refer siano pari al 4%, il numero dei casi che dovranno eseguire gli ABR saranno circa 40 per mil e nati (fra essi vi saranno 1-1,5 casi con conferma di ipoacusia). Calcolando un costo tariffario di ciascun ABR diagnostico infantile pari a 35 euro, il costo complessivo di questa fase è pari Questi costi non sono in realtà costi aggiuntivi, ma una razionalizzazione di quel i attua- li. Dato che il numero di bambini con problemi di ipoacusia non si modifica, ma si anticipa solo la diagnosi, calcolando una prevalenza del e ipoacusie congenite attorno al '1-1,5‰, il costo complessivo del o screening è di circa 6-7.000 € per ogni bambino diagnosticato
sordo entro i primi sei mesi di vita. I costi che seguono la diagnosi (protesizzazione, fol-
low-up .) sono gli stessi; si ha invece una riduzione dei costi di riabilitazione in quanto, an- che se questa viene anticipata, richiede nel tempo costi minori, dato il marcato migliore risul- tato del 'intervento precoce sul o sviluppo del linguaggio. 10.3 I costi sociali
Una ricerca condotta dal 'Istituto Italiano di Medicina Sociale ha dimostrato che i costi sociali della sordità profonda prelinguale per singolo alunno del e scuole materne e ele- mentari nel 2003, per il solo sostegno scolastico, era pari a circa 19.000 €, mentre per la scuola media di I e II grado a circa 14.000 € al 'anno. Se calcoliamo che gli alunni con handicap uditivo nel 'anno scolastico 2002/2003 erano in Italia 5.851, la spesa annuale complessiva è stata pari a 99.945.887 €. A questi si devono aggiungere i costi per l'istruzione logopedica ed in seguito i costi previdenziali. Ben si comprende come lo screening neonatale del a sordità congenita, ac- compagnato da una diagnosi corretta e da una terapia appropriata, venga considerato a li- vel o internazionale un intervento sanitario che presenta uno dei migliori rapporti co-
11. CONSIDERAZIONI CONCLUSIVE
Malgrado le raccomandazioni del e Società scientifiche a livel o internazionale e na- zionale e l'impegno del e Società italiane di pediatria, neonatologia, otorinolaringoiatria, audiologia e foniatria, lo screening uditivo neonatale non è ancora stato definitivamente in- serito nei livel i essenziali di assistenza, anche se alcune regioni italiane lo hanno reso ob- bligatorio e altre hanno iniziato l'iter legislativo. Il Piano sanitario nazionale 2011-2013, fra le priorità da realizzare nel Triennio, indica l'estensione del o screening audiologico neonatale della sordità congenita per raggiungere almeno il 90% dei neonati, condividendo un documento tecnico di indirizzo che definisca le modalità del o screening ed il percorso del paziente affetto da sordità. Il programma di screening, diagnosi e terapia appropriata del e sordità congenite de- ve prevedere: a - Screening audiologico a tutti i neonati. Per ottenere una copertura della popolazione pari al 95%, il metodo migliore è quel o di effettuare il test di screening prima del a dimissione dal punto nascita, il programma di screening deve essere concluso entro il primo mese di vita (i soggetti "refer" al a dimissione eseguono un retest dopo circa 2- b - Diagnosi audiologica entro i tre mesi di vita. c - Adeguato e puntuale programma di intervento riabilitativo entro i 6 mesi. d - Se indicato, impianto cocleare entro 1-2 anni. e - Coordinamento dei servizi che a livello territoriale e specialistico devono occuparsi del piccolo paziente con un puntuale coinvolgimento dei pediatri di famiglia. f - Formazione continua, possibilmente congiunta ECM di tutti gli operatori coinvolti. g - Informazione corretta ed educazione sanitaria a tutti i componenti del a famiglia con eventuale supporto psicologico. h - Il sostegno alle famiglie costituisce il cardine per il successo del a riabilitazione (75). i - Integrazione funzionale dei tre livel i di intervento e informatizzazione del 'intero mo- del o operativo. l - Aggiornamento del nomenclatore tariffario, tenendo conto del e acquisizioni tecnolo- giche e del a fascia economica del e famiglie. m - Elaborazione e approvazione di Linee guida nazionali che definiscano con chiarezza: le modalità di esecuzione del o screening audiologico neonatale, la conferma del a diagnosi, il percorso del paziente affetto da sordità, la sorveglianza audiologica del e età successive, l'appropriatezza del 'impianto cocleare, compresi gli standard qualita- tivi e quantitativi dei Centri di III livel o e del e strutture dove eseguire l'impianto co-
Bibliografia
1. American Academy of Pediatrics, Task Force on Newborn and Infant Hearing. New-
born and infant hearing loss: detection and intervention. Pediatrics. 1999; 103:527- 2. Joint Committee on Infant Hearing. Year 2000 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programme. Pediatrics. 2000; 106:798-817 3. American Academy of Pediatrics, Committee on Practice and Ambulatory Medicine and Section on Otolaryngology and Bronchoesophagology. Hearing Assessment in Infants and Children: Recommendations Beyond Neonatal Screening. Pediatrics. 2003; 111:436-440 4. American Academy of Pediatrics, US Preventive Service Task Force. Universal Screening for Hearing Loss in Newborns: Recommendation Statement Pediatrics. 2008; 122:143-148 5. European Consensus Development Conference on Neonatal Hearing Screening. 15- 16 May 1993:1-24 6. Yoshinaga-Itano C, Sedey A, Coulter D, Mehl A. Language of early and later identi- fied children with hearing loss. Pediatrics 1998; 102:1161-1171 7. Sharma A, Tobey E, Dorman M, Bharadwaj S, Martin K, Gil ey P, Kunkel F. Central auditory maturation and babbling development in infants with cochlear implants. Arch Otolaryngol Head Neck Surg. 2004;130(5):511-6. 8. Sharma A, Campbel J.A sensitive period for cochlear implantation in deaf children. J Matern Fetal Neonatal Med. 2011;24 Suppl 1:151-3. 9. Coplan J. Deafness: ever heard of it? Delayed recognition of permanent hearing loss. Pediatrics. 1987; 79:206-213 10. Watkin PM, Baldwin M, Laoide S. Parental suspicion and identification of hearing im- pairment. Arch Dis Child. 1990; 65:846-850 11. Harrison M, Roush J. Age of suspicion, identification, and intervention for infants and young children with hearing loss: a national study. Ear Hear 1996; 17:55-62 12. Kittrel AP, Arjmand EM. The age of diagnosis of sensorineural hearing impairment in children. Int J Pediatr Otorhinolaryngol. 1997; 40:97-106 13. Servil e MN, Demanez L, Demanez JP. Diagnosis of hearing impairment: factors of delay. Acta oto-rhino-laryngol belg. 2004; 58:53-59 14. Harrison M, Roush LJ, Wal ace J. Trends in age of identification and intervention in infants with hearing loss. Ear Hear. 2003; 24(1):89-95 15. Fortnum H, Davis A. Epidemiology of permanent childhood hearing impairment in Trent Region 1985-1993. Br J Audiol. 1997; 31:409-446 16. Davis A, Hind S. The newborn hearing screening programme in England. Int J Pediatr Otorhinolaringol. 2003; 6751:193-196 17. Fortnum H, Sommerfield Q, Marshall D, Davis A, Bamford J. Prevailing of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ. 2001; 18. Morton CC, Nance WE. Newborn Hearing Screening - A Silent Revolution. N Engl J Med. 2006; 354;20:2151-2164 19. Pelosi G, Hatzopoulos S, Chierici R, Vigi V, Martini A. Evaluation of a linear TEOAE protocol in hearing screening of neonates: feasibility study]. Acta Otorhinolaryngol Ital. 1998 Aug;18:213-7. 20. Hatzopoulos S, Tsakanikos M, Grzanka A, Ratynska J, Martini A. Comparison of neonatal transient evoked otoacoustic emission responses recorded with linear and QuickScreen protocols. Audiology. 2000;39:70-9. 21. Hatzopoulos S, Pelosi G, Petruccel i J, Rossi M, Vigi V, Chierici R, Martini A. Efficient otoacoustic emission protocols employed in a hospital-based neonatal screening pro- gram. Acta Otolaryngol. 2001;121:269-73. 22. Jedrzejczak WW, Hatzopoulos S, Martini A, Blinowska KJ. Otoacoustic emissions la- tency difference between ful -term and preterm neonates. Hear Res. 2007 ;231:54-62 23. Ciorba A, Hatzopoulos S, Busi M, Guerrini P, Petruccel i J, Martini A. The universal newborn hearing screening program at the University Hospital of Ferrara: focus on costs and software solutions. Int J Pediatr Otorhinolaryngol. 2008 ;72:807-16. 24. Hatzopoulos S, Petruccel i J, Ciorba A, Martini A. Optimizing otoacoustic emission protocols for a UNHS program. Audiol Neurootol. 2009;14:7-16. 25. De Capua B, Costantini D, Martufi C, Latini G, Gentile M, De Felice C. Universal neo- natal screening: The Siena (Italy) experience on 19,700 newborns. Early Hum Dev 26. Bubbico L, Rosano A, Spagnolo A, La prevalenza della sordità profonda prelinguale in Italia. La Sordità infantile. Istituto Italiano di medicina sociale. 27. Kral A, O'Donoghue GM. Profound Deafness in Childhood. N Engl J Med. 2010; 363;15:1438-1450 28. Declau F, Boudewyns AU, van den Ende J, et al. Etiological and audiological evalua- tions after universal neonatal hearing screening: analysis of 170 referred neonates. Pediatrics. 2008; 121:1119-1126 29. Denoyel e F, Marlin S, Weil D, et al. Clinical features of the prevalent form of child- hood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counsel ing, Lancet. 1999; 353:1298-1303 30. Lefebvre PP, van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene. Brain Res Rev. 2000; 32:159-162 31. Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non- syndromic sensorineural deafness. Nature. 1997; 387:80-83 32. Gasparini P, Rabionet R, Barbujani G, et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur J Hum Genet. 2000; 8:19-23 33. Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyel e F, Waligora J, Muel er- Malesinska M, Pollak A, Ploski R, Murgia A, Orzan E, Castorina P, Ambrosetti U, Nowakowska-Szyrwinska E, Bal J, Wiszniewski W, Janecke AR, Nekahm-Heis D, Seeman P, Bendova O, Kenna MA, Frangulov A, Rehm HL, Tekin M, Incesulu A, Dahl HH, du Sart D, Jenkins L, Lucas D, Bitner-Glindzicz M, Avraham KB, Brown- stein Z, del Castil o I, Moreno F, Blin N, Pfister M, Sziklai I, Toth T, Kel ey PM, Cohn ES, Van Maldergem L, Hilbert P, Roux AF, Mondain M, Hoefsloot LH, Cremers CW, Löppönen T, Löppönen H, Parving A, Gronskov K, Schrijver I, Roberson J, Gualandi F, Martini A, Lina-Granade G, Pal ares-Ruiz N, Correia C, Fialho G, Cryns K, Hilgert N, Van de Heyning P, Nishimura CJ, Smith RJ, Van Camp G. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945-57. 34. Hilgert N, Huentelman MJ, Thorburn AQ, Fransen E, Dieltjens N, Muel er-Malesinska M, Pol ak A, Skorka A, Waligora J, Ploski R, Castorina P, Primignani P, Ambrosetti U, Murgia A, Orzan E, Pandya A, Arnos K, Norris V, Seeman P, Janousek P, Feldmann D, Marlin S, Denoyel e F, Nishimura CJ, Janecke A, Nekahm-Heis D, Martini A, Men- nucci E, Tóth T, Sziklai I, Del Castil o I, Moreno F, Petersen MB, Iliadou V, Tekin M, Incesulu A, Nowakowska E, Bal J, Van de Heyning P, Roux AF, Blanchet C, Goizet C, Lancelot G, Fialho G, Caria H, Liu XZ, Xiaomei O, Govaerts P, Grønskov K, Host- mark K, Frei K, Dhooge I, Vlaeminck S, Kunstmann E, Van Laer L, Smith RJ, Van Camp G. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet. 2009;17:517-24. 35. Gualandi E, Ravani A, Berto A, Burdo S, Trevisi P, Ferlini A, Martini A, Calzolari E. Occurrence of del(GIB6-D13S1830) mutation in Italian non-syndromic hearing loss patients carrying a single GJB2 mutated al ele. Acta Otolaryngol Suppl. 2004;552:29- 36. Gualandi F, Ravani A, Berto A, Sensi A, Trabanel i C, Falciano F, Trevisi P, Mazzoli M, Tibiletti MG, Cristofari E, Burdo S, Ferlini A, Martini A, Calzolari E. Exploring the clinical and epidemiological complexity of GJB2-linked deafness. Am J Med Genet. 37. Berto A, Pel ati D, Castiglione A, Busi M, Trevisi P, Gualandi F, Ferlini A, Martini A. Audiological profiles and gjb2, gjb6 mutations: A retrospective study on genetic and clinical data from 2003 to 2008. Audiol. Med 7: 93-105, 2009 38. Gorlin RJ, Toriel o HV, Cohen MM. Hereditary hearing loss and its syndromes. Oxford University Press. 1995; 1-457 39. Bom SJH, Kunst HPM, Huygen PLM, et al. Non-syndromal autosomal dominant hearing impairment: ongoing phenotypical characterization of phenotypes. Br J Audiol. 1999; 33:335-348 40. Fischel-Ghodsian N, Prezant TR, Chaltraw WB, et al. Mitochondrial gene mutation is a significant predisposing factor in aminoglycoside ototoxicity. Am J Otolaryngol. 1997; 18:173-178 41. Martini A. Genetica del a funzione uditiva normale e patologica. Ed. Omega, Torino. 42. Kimberling WJ, Weston MD, Möl er C, Davenport SL, Shugart YY, Priluck IA, Martini A, Milani M, Smith RJ. Localization of Usher syndrome type II to chromosome 1q. Genomics. 1990;7:245-9. 43. Kimberling WJ, Weston MD, Pieke Dahl S, Kenyon JB, Shugart YY, Mol er C, Daven- port SL, Martini A, Milani M, Smith RJ. Genetic studies of Usher syndrome. Ann N Y Acad Sci. 1991;630:167-75. 44. Kimberling WJ, Weston MD, Möl er C, van Aarem A, Cremers CW, Sumegi J, Ing PS, Connol y C, Martini A, Milani M, et al. Gene mapping of Usher syndrome type IIa: lo- calization of the gene to a 2.1-cM segment on chromosome 1q41. Am J Hum Genet. 1995;56:216-23 45. Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, Smith RJ, Sumegi J, Kimberling WJ. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66:1199-210. 46. Ciorba A, Schrott-Fisher A, Berto A, Glueckert R, Janecke A, Martini A. Histopa- thological and neuroradiological features of Usher syndrome type II. B-ENT. 47. Martini A, Calzolari F, Sensi A. Genetic syndromes involving hearing. Int J Pediatr Otorhinolaryngol. 2009;73 Suppl 1:S2-12. 48. Zadro C, Ciorba A, Fabris A, Morgutti M, Trevisi P, Gasparini P, Martini A. Five new OTOF gene mutations and auditory neuropathy. Int J Pediatr Otorhinolaryngol. 2010 49. Vozzi D, Aaspõl u A, Athanasakis E, Berto A, Fabretto A, Licastro D, Külm M, Testa F, Trevisi P, Vahter M, Ziviel o C, Martini A, Simonelli F, Banfi S, Gasparini P. Molec- ular epidemiology of Usher syndrome in Italy. Mol Vis. 2011;17:1662-8. 50. Busi M, Castiglione A, Taddei Masieri M, Ravani A, Guaran V, Astolfi L, Trevisi P, Ferlini A, Martini A. Novel mutations in the SLC26A4 gene. Int J Ped Otol. 2012; 51. Canningham M, Cox EO. Hearing assessment in infant and children: Recommenda- tions beyond neonatal screening. Pediatrics. 2003; 111:436-440 52. Barbi M, Binda S, Primache V, Luraschi C, Corbetta C. Diagnosis of congenital cy- tomegalovirus infection by detection of viral DNA in dried blood spots. Clin Diagn Vi- rol. 1996; 6:27-32 53. Barbi M, Binda S, Caroppo S, Primache V. Neonatal screening for congenital cy- tomegalovirus infection and hearing loss. J Clin Virol. 2006; 35:206-209 54. Stehel EK, Shoup AG, Owen KE, et al. Newborn hearing screening and detection of congenital cytomegalovirus infection. Pediatrics. 2008; 121:970-975 55. Barbi M, Binda S, Caroppo S, Ambrosetti U, Corbetta C, Sergi P. A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr Infect Dis J. 2003; 22:39-42 56. Kimberlin DW, Lin CY, Sauchez PJ, et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous sys- tem: a randomized, control ed trial. J Pediatr. 2003; 143:16-25 57. Lanari M, Lazzarotto T, Venturi V, et al. Neonatal cytomegalovirus blood load and risk of sequelae in symptomatic and asymptomatic congenital y infected newborns. Pedi- atrics. 2006; 117:76-83 58. Ciorba A, Bovo R, Trevisi P, Bianchini C, Arboretti R, Martini A. Rehabilitation and outcome of severe profound deafness in a group of 16 infants affected by congenital cytomegalovirus infection. Eur Arch Otorhinolaryngol. 2009;266:1539-46. 59. Pass RF, Zhang C, Evans A, et al. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med. 2009; 360:1191-1199 60. Peltola H, Roine I, Fernández J et al. Hearing Impairment in Childhood Bacterial Meningitis Is Little Relieved by Dexamethasone or Glycerol. Pediatrics. 2010; 125:e1 61. Worsøe L, Cayé-Tomasen P, Brandt CT, et al. Factors Associated with the Occur- ence of Hearing Loss after Pneumococcal Meningitis. Clin Infect Dis. 2010; 51:8 62. Kopelovich JC, Germil er JA, Laury AM, et al. Early Prediction of Postmeningitic Hearing Loss in Children Using Magnetic Resonance Imaging. Arch Otolaryngol. Head Neck Surg. 2011; 137. 441-447 63. Reefhuis J, et al. Risk of Bacterial Meningitis in Children with Cochlear Implants, USA 1997-2002. Engl J Med. 2003; 349:435-45 64. Biernath KR, Reefhuis J, Whitney CG, et al. Bacterial Meningitis Among Children Eith Cochlear Implants Beyond 24 Months After Implantation. Pediatrics. 2006; 117, 2 65. Rubin LG, Papsin B, Committee on Infectious Diseases and Section on Otolaryngol- ogy Head and Neck Surgery. Pediatrics. 2010; 126;381-391 66. Universal Screening for Hearing Loss in Newborns: US Preventive Services Task Force. Recommendation Statement. Pediatrics. 2008, 122:143-8.1 67. Sergi P, Pastorino G, Ravazzani P, Tognola G, Grandori F. A hospital based univer- sal neonatal hearing screening programme using click-evoked otoacoustic emis- sions.Scand Audiol Suppl. 2001;(52):18-20. 68. Bubbico L, Bartolucci MA, Broglio D. The newborn hearing screening in Italy. Letter to editor. Ital J Pediatr. 2005; 31:290-292 69. Bubbico L, Greco A, Tognola G, Grandori F. Universal newborn hearing screening programs in Italy: survey of year 2006. Acta Otolaryngol. 2008; 22:0-23 70. Pastorino G, Sergi P, Mastrangelo M, et al. The Milan Project: A newborn hearing screening programme. Acta Paed. 2005; 94:458-463 71. Suppiej A, Rizzardi E, Zanardo V, et al. Reliability of hearing screening in high-risk neonates: Comparative study of otoacoustic emission, automated and conventional auditory brainstem response. Clin Neurophysiol. 2007; 118:869-876 72. Weichbold V, Nekahm-Heis D, Welzl-Muel er K. Universal Newborn Hearing Screen- ing and Postnatal Hearing Loss. Pediatrics. 2006; 117:631-6 73. American Academy of Pediatrics, Joint Committee on Infant Hearing. Year 2007 posi- tion statement: principles and guidelines for early hearing detection and intervention program. Pediatrics. 2007; 120, 4:898-921 74. Pisacane A, Arslan E, Auletta G, et al. Lo screening neonatale dei disturbi permanenti del 'udito. Prospettive in Pediatria. 2009; 39, 156:193-199. 75. White KR. Early hearing detection and intervention programs: Opportunities for ge- netic services. Am J Med Genet. 2004; 130A:29-36 76. Cutler J, Lenzi G, Berrettini S, Martini A, Martinel i S. How to motivate newborn hear- ing screening in the absence of a national programme: a col aboration between parents fessionals. J Matern Fetal Neonatal Med. 2012 Oct;25 Suppl 4:106-7.

Fig. 1 - Cause di sordità
Emorragia cerebrale
Meningite batterica
Incompatibilità AB0
Sordità sindromiche
Sindrome fetale da alcool
Patologie cromosomiche
Sordità familiare
Fig. 2 - La diagnosi eziologica del e sordità congenite
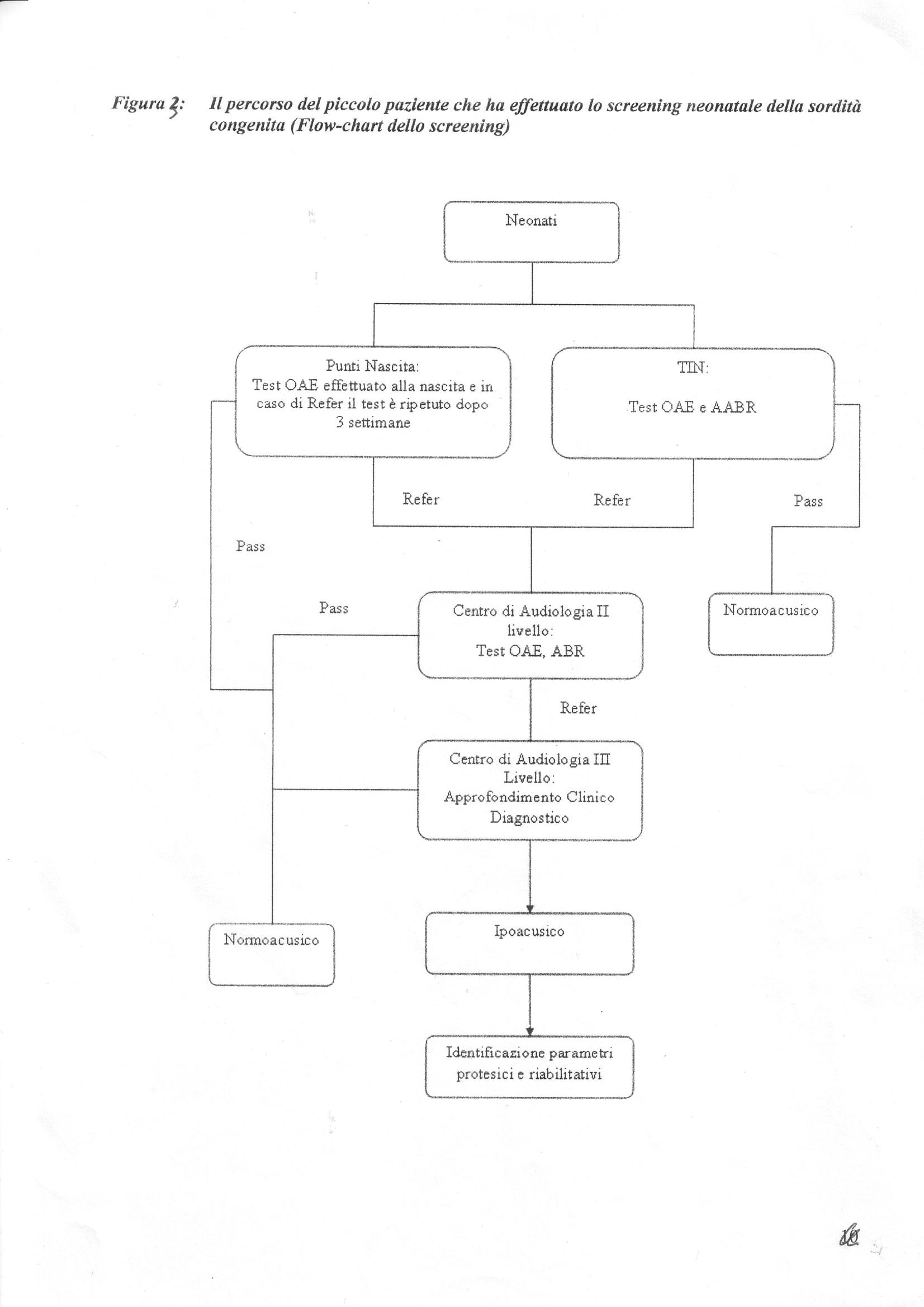
Fig. 3 - Il percorso del piccolo paziente che ha effettuato lo screening neonatale
del a sordità congenita (Flow-chart del o screening)
Source: http://www.orlpadova.it/Pers/ORLPADOVA/userfiles/files/screening%20neonatale_%20cart%20Audiologia.pdf
Microsoft word - mps_procedure_guidelines final 12.doc
Procedure Guidelines for Radionuclide Myocardial Perfusion Imaging with Single-Photon Emission Computed Adopted by the British Cardiac Society, the British Nuclear Cardiology Society, and the British Nuclear Medicine Society Writing Group: P Arumugam; M Harbinson, E Reyes, N Sabharwal, C Tonge, SR Underwood Advisory Group: Andrew Kelion
Natural high
COPYRIGHT 2006 SCIENTIFIC AMERICAN, INC. COPYRIGHT 2006 SCIENTIFIC AMERICAN, INC. The brain produces its own "marijuana" to protect neurons, and researchers hope to exploit it to ease anxiety, obesity and addiction COPYRIGHT 2006 SCIENTIFIC AMERICAN, INC. hemically speaking, we are all potheads. ling appetite, and in the cerebellum, which gov- Raphael Mechoulam of Hebrew Uni-