Ame.nd.edu
Polymeric Micelles – The Future of Oral Drug DeliveryDepartment of Chemical and Biomolecular Engineering University of Notre Dame, Notre Dame, IN 46556 Abstract
This work examines current advancements in polymeric micelles as a method for oral
delivery of poorly water-soluble drugs. The oral route presents several barriers to drug
delivery that the chosen vesicle must overcome. Polymeric micelles have several
physical properties, including molecular weight and copolymer block composition, which
can be tailored to alter the vesicle structure and overcome these barriers. Examination of
current research demonstrates the ability of polymeric micelles to respond to external
stimuli, such as pH, allowing for controlled release of encapsulated drugs in the
gastrointestinal tract. Lastly, with patients preferring the oral drug delivery route to the
intravenous delivery route, it was shown that polymeric micelles can achieve the same
desired pharmacological dose via either delivery method. These factors make polymeric
micelles appear to be a viable option for future oral drug delivery applications.
1. Introduction
1.1 Clinical Relevance
Advances in current medicine have made it necessary to develop novel drug delivery systems (DDSs). Medication can be administered in several ways, with 112 routes for administration approved by the FDA [1]. Currently, it is rare for a pharmaceutical product to enter the market without its own specific delivery system [2]. The newest products in the market are biologics, such as peptides and proteins, because of the ability to provide highly selective, effective, and potent action in treatment of multiple diseases [3]. However, most biologic drugs are administered intravenously because of the multiple barriers posed by the gastrointestinal tract (GIT). These barriers include physiochemical conditions of the GIT, low levels of penetration across the transepithelial membrane, and poor bioavailability due to low drug solubility in the GIT lumen [4]. Oral DDSs are widely used for non-biologic medical treatments because it is the easiest and most convenient method of delivery when repeated or routine administration is required [5]. This route is favored because of increased patient compliance, less stringent quality control, improved safety, lower costs, and no requirement for trained professionals to administer injections [4]. A patient with multiple sclerosis, for which the only current treatment is a weekly injection, said "If I could take a pill, I almost wouldn't mind the disease," eliminating the syringes "would make it a lot more tolerable" [6]. These benefits lead to the fact that oral products account for nearly 70% of the value in the US pharmaceutical market and 60% of the DDSs used [2]. With such a large market
share currently held by oral DDSs, research has been focused on oral DDSs that address the three barriers limiting biologic drugs.
Polymeric micelles (PMs) have become an area of focus because they can easily
be modified to address the current limitations for oral bioavailability. Namely, they are small in size which favors transport across the mucosal epithelium and the micellar structure can be modified to provide enhanced stability in various environments [3]. PMs also have a critical advantage since the combined hydrophobic/hydrophilic structure helps improve the solubility of poorly water-soluble drugs. With as high as 40% of potential drugs that pass through screening rejected from formulation development on the basis of poor solubility, this structural advantage is beneficial [7]. Since PMs have many properties that allow them to overcome the three main barriers currently restricting oral administration of biologics, they are an excellent candidate for future novel drug delivery systems. 1.2 Gastrointestinal Tract Anatomy
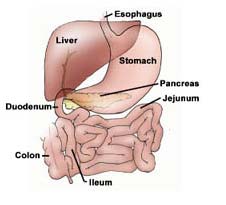
The anatomy of the GIT (Figure 1) is the
most important factor in determining the necessary physiochemical characteristics of a material for effective oral delivery. In oral DDSs, the drug must be absorbed through the intestinal membrane which presents both morphological barriers, such as the mucosal layer and microvilli, and physiological barriers, such as a wide pH range, digestive enzymes, and specific transport mechanisms [9]. Drug molecules are thought to absorb across the intestinal membrane into the bloodstream via Figure 1. A schematic diagram of three main mechanisms (Figure 2). These the anatomy of the human mechanisms are the paracellular passage of small gastrointestinal tract [8]. particles, in the micron size range, through small gaps between intestinal epithelial cells, transcellular passive diffusion through the cell membrane, and receptor-mediated transcytosis via endocytosis [9]. Polymeric micelles are thought to absorb in the intestine and accumulate in the bloodstream via transcytosis.
Figure 2. Three main mechanisms for drug absorption across the intestinal membrane. (A) Paracellular passage through small gaps; (B) Transcellular passive diffusion through cell membrane; (C) Receptor-mediated endocytosis via transcytosis [9].
In the GIT, particle uptake occurs mainly at the M-cells of Peyer's patches (PPs). PPs are more accessible than other epithelial surfaces in the GIT because the M-cells have a smaller glycocalyx layer. The glycocalyx is a 400-500 nm layer that contains negatively charged mucosal molecules as well as the pancreatic and glycoprotein enzymes involved in digestion [4]. The M-cells have more open space on the apical membrane and lack fully developed microvilli which allows for particles in the GIT lumen to localize at these areas and be transcytosed (Figure 3) [5]. Various particle properties including size, hydrophobicity, and charge affect the extent of particle uptake. Decreasing particle size, which increases surface area, leads to better uptake of particles [9]. With sizes on the order of 10 nm, PMs are favored for uptake and absorption in most tissues [7]. This small size allows PMs to be internalized by cells more efficiently since they can reach very small vascular vessels as well as accumulate in cells with leaky, compromised vasculature, like cancer cells [10]. A more hydrophobic surface has also been shown to absorb favorably [9]. Hydrophobic molecules generally have poor wettability by water, but the native surfactants in the GIT lumenal fluids can improve the wetting of hydrophobic surfaces, leading to Figure 3. Electron microscopy of the better particle uptake [11]. Charge has no Peyer's patch epithelium showing an clear effect on particle uptake because neutral M-cell and its neighboring absorptive or positively charged particles exhibit a higher cell [5]. binding to the epithelia but negatively charged particles have bioadhesive properties which favor the transcytosis process [9]. As these factors all play important roles in the oral uptake process, polymeric micelles are advantageous because these physical properties can be modified by altering the polymers used in formation.
Lastly, the pH of the GIT can play a significant role in both the charge and shape
of the polymer. The GIT exhibits a wide pH range that depends on multiple factors including the fed state (Table 1), age, pathiophysiological conditions, and concurrent drug therapies [11]. This wide pH range is important since the presence of weakly acidic or basic functional groups in the backbone of the polymer chain causes multiple polymers to exhibit a pH-dependent behavior. Polymers exhibiting pH-dependent behavior can swell or shrink in a controllable manner from changes in the porosity of the polymer network due to pH fluctuations [2]. By understanding the pH of the GIT, PMs can be developed that exhibit preferential release of the encapsulated drug in the intestinal tract where the uptake of the drug is more likely.
Table 1. pH values for various locations in the GIT at different fed states [11]
2. Polymeric Micelles
2.1 Micellar Structure
Polymeric micelles are formed from amphiphilic copolymers that have a
hydrophilic block and a hydrophilic block. The choice of amphiphilic copolymer architecture can result in several different possible micellar morphologies (Figure 4) [10].
Figure 4. Various micellar morphologies that can be formed spontaneously in aqueous solution from different copolymer architectures [10].
All PM morphologies are driven towards spontaneous formation in aqueous solution by a reduction in the system free energy [10]. This reduction occurs because of an entropic gain from solvation as the hydrophobic polymer segment is removed from the aqueous environment and aggregates [12]. The hydrophobic segment aggregates via Van der Waals interactions while the hydrophilic segment reestablishes a hydrogen-bonded network with water [7]. During this process, the copolymers give rise to a unique structure – a hydrophobic core stabilized by a hydrophilic corona [12]. The hydrophobic core efficiently incorporates poorly water soluble drugs and the hydrophilic corona makes the drugs water soluble since it interacts with the aqueous solution, shielding the core from interactions with water [10]. The ability to manipulate the chemistry of these two block regions allows for easy manipulation of the micellar structure to yield an efficient DDS. In determining the polymer for use in drug incorporation, it is important to match the polarity of the hydrophobic core to the solubility of the drug. The solubility characteristics of the drug determine its loading region to be Figure 5. Possible patterns of micellar drug
preferentially located inside the core, in
association based on hydrophobic (black) to
the corona, or within the core/corona hydrophilic (white) area. 1) Completely
interface (Figure 5) [9]. This water-soluble only adsorbed in corona. 2-4)
core/corona interface depends on the Intermediate cases of hydrophobic/philic
micellar morphology with different drugs that adsorb at core/corona interface.
morphologies achievable depending on
5) Completely insoluble drug adsorbs into
the amphiphilic copolymer molecular the micellar core [13].
weight, physical state, and composition as well as the solution pH, solvent, polymer concentration, and ionic strength [10]. Of these properties, focus is given to the molecular weight, polymer concentration, and composition since these characteristics are easiest to modify and control during PM formation.
In terms of solution effects, PMs only form spontaneously once a given polymer
concentration is reached, similar to surfactant micelles. This concentration is known as the critical micellar concentration (CMC); below the CMC the polymer exists as unimers, or single units, and above the CMC, unimers still exist, but they also spontaneously self-assemble into micelles [10]. A minimum critical solution temperature must also be overcome for spontaneous self-assembly of the unimers into PMs [10]. These two conditions play important roles in the stability of the PMs in vivo. Typical physiological temperatures are above the minimum micellar temperature for most polymers, but upon oral administration a DDS can be severely diluted by the GIT fluids. For this reason, the CMC is an important factor to make sure dosage concentrations remain above the CMC when ingested, preventing early release from spontaneous degradation of the PMs [9].
Compared to typical surfactants such as sodium dodecyl sulfate (SDS), PMs are much less sensitive to these dilution effects because of the lower CMCs. For example, in aqueous solution SDS has a CMC of 2300 mg/L [14] whereas a hydrophobically modified polysaccharide PM has a CMC of 5 mg/L [9]. This nearly 500 fold decrease in CMC makes PMs suitable for avoiding dilution effects in the GIT fluid [10]. If the PM will be diluted by a know amount of fluid or the amount can be estimated, then a safety factor can be added such that the polymer concentration in solution ensures no spontaneous degradation and undesired drug release from the PM due to dilution below the CMC.
Another factor that plays an important role in the micelle structure and the CMC
is the composition of the block copolymer. Composition affects that play a large role in structure and stability are the molecular weight, i.e. chain length, core structure, and cross-linking of the block segments. If the hydrophilic block is too long, the copolymers will not spontaneously form micelles in solution and will only exist as unimers. If the hydrophobic block is too long, the copolymers will spontaneously form non-micellar structures, like rods and lamellae [7]. The lengths of these respective chains also affect the CMC of the polymer. If the hydrophilic chain is kept constant and the hydrophobic chain is lengthened, the CMC will decrease, increasing micelle stability. On the other hand, keeping the hydrophobic chain length constant and increasing the hydrophilic block length increases the CMC [9]. The core structure of the PM can be amorphous or crystalline depending on the glass transition temperature of the hydrophobic block. If the glass transition temperature exceeds physiological temperature the core is stabilized due to restricted motion of the polymer segments yielding better kinetic stability upon dilution. A crystalline core can also lower the diffusion of a drug from the core compared to amorphous core structures [12]. Cross-linking can further enhance stability upon dilution. Cross-linking generates stable bonds in the water-soluble corona that can resist shear forces and dilution because of the immobile but permeable cross-linked surface [12]. All these beneficial properties can be tailored using polymer chemistry to determine what block copolymers should be used. 2.2 Polymers Used
Numerous polymers can be used to form micelles, with variations in the
hydrophilic and hydrophobic blocks generating an expansive library for possible drug delivery vehicles. Since these PMs are going to be delivering a drug to the body and then digested by the body, they must be an FDA approved device. These requirements limit the choices for the various blocks since it is easier to develop new technology using polymers already used in previously FDA approved devices. In general for pharmaceutical applications, the amphiphilic polymers should exhibit biocompatibility and non-toxicity [10].
Multiple choices for the hydrophilic block polymer exist. The most commonly
used polymer is poly(ethylene glycol) (PEG) because it is inexpensive, has low toxicity, and is a good steric protector for many biologically active macromolecules. It is also already used in devices approved by most regulatory agencies for internal applications [7]. Even though PEG is not biodegradable, it is easily removed from the body via the excretion pathways if under a molecular weight of 15 kDa [10]. Nearly all current
research focuses on PEG chains of various molecular weights, but other polymers can be used. Those that are currently generating interest include poly(N-vinyl-2-pyrrolidone) and polyvinyl alcohol. They both have biocompatibility and water-solubility similar to PEG [10]. If temperature or pH sensitivity is required, poly(N-isopropylacrylamide) (PNIPA) can be used [7].
Several polymers meet the requirements of pharmaceutical application for the
hydrophobic core forming block. Most of these polymers are of the poly(ester) and
poly(amino acid) families [15]. The most commonly used ones in research include
poly(propylene oxide) (PPO), poly(caprolactone) (PCL), poly(L-lactic acid) (PLLA),
poly(lactic-co-glycolic acid) (PLGA), poly(aspartic acid) (PAsp), poly(glutamic acid)
(PGlu), poly(L-lysine) (PLys), and various poly(acrylates) [7,10,15]. The main variation
among these is whether or not charge is desired for incorporating the drug and the bond
stability for controlled release properties. Most of these polymers also break down into
unimers that already exist in the body and as such are commonly used in FDA approved
devices. One example is PLGA which is both biocompatible and biodegrable, breaking
down to lactic acid and glycolic acid, both byproducts of other metabolic pathways in the
body. It is these various options for the two blocks that make PMs tailorable to a given
drug and a viable option for future DDSs.
3. Drug Delivery
3.1 Modes of Drug Release
The rate of drug release from PMs is a very important control for oral drug
delivery. Compared to intravenous administration, which results in significant concentration of a drug immediately after injection and a lower than required threshold of dose near the end of the dosing period, oral delivery can achieve prolonged and continuous exposure at a lower and safer concentration that is still pharmacologically effective. This lower dose from oral delivery can avoid many toxic side effects and improve the efficacy of the drug [12]. The release rate is controlled by several factors including chemical structure of the copolymer, the incorporated drug and its localization, and micelle preparation method, but the main factor depends on the hydrolysable chemical bond formed between the drug and polymer as well as the cross-linking bonds in the polymer [15]. A stable bond is less likely to degrade and enhances micelle protection of the drug. The typical polymers used in the hydrophobic block have either ester bonds or amide bonds (amino acids). When undergoing only hydrolysis at physiological conditions with no enzymes present these bonds exhibit very high stability (Table 2). When an ester bond or amide bond is cleaved, one resultant product is an acid which lowers the pH and increases degradation. This increased degradation of the bonds is a result of the self-catalysis of hydrolysis which can be catalyzed by either an acid or a base [16].
Table 2. Bond half-life for various polymer classes undergoing hydrolysis at physiological conditions with no enzymes present [16].
Two major pathways exist by which the encapsulated drug is released from the
micellar core block. These pathways are micellar dissociation followed by drug cleavage from the unimer and drug cleavage within the micelle followed by diffusion out of the delivery system (Figure 6) [15]. In each of these pathways, there are various ways to control the cleavage. For micellar dissociation, three mechanisms exist by which the degradation occurs (Figure 7). Each mechanism depends on the various types of bonds in the polymer network and the relative stability of the bond [16]. For drug cleavage followed by diffusion, the release depends on the chemical conjugation of the drug to the hydrophobic polymer [15]. PMs are advantageous because of the variation available to alter the structure according to the desired requirements. Enhancing hydrophobicity and rigidity of the core restricts water penetration leading to sustained or delayed drug release from the carrier system. A glassy core at physiological temperature, cross-linking, and hydrophobic interactions or hydrogen bonding with the drug lowers micellar dissociation, drug diffusion, and thus overall drug release. An instant release of drug can be achieved by introducing a hydrophilic or stimulus responsive group to the core structure [15].
Figure 6. Two mechanisms for drug release from micelle-forming block copolymer-drug conjugate [15].
Figure 7. Three mechanisms for solid polymer degradation. I) Cleavage of cross-links between polymer chains. II) Chemical cleavage/transformation of polymer backbone side chains. III) Direct cleavage of polymer backbone [16].
Examination of various chemical conjugations of drugs to the hydrophobic chain
has been investigated to determine the behavior in different physiological pH ranges. Doxorubicin (DOX) is a widely used anticancer agent and is mildly water-soluble so it could not be sufficiently loaded into PMs by physical addition of DOX to a polymer solution above the CMC [17]. By chemically conjugating the primary amino group of DOX to the terminal hydroxyl group of PLGA it was shown that a more sustained release profile of DOX was achieved compared to physically encapsulated DOX in PEG-PLGA polymeric micelles. The only problem observed with this linkage was that it could not be cleaved under physiological conditions and the released DOX was still conjugated to PLGA oligomers [18]. To eliminate this problem, acid-cleavable linkages, a hydrazone bond and a cis-aconityl bond, were used to conjugate DOX to the terminal end of PLLA in a PLLA-PEG copolymer. The release profiles for these two bonds were studied over physiological pH ranges for short term release of DOX (Figure 8). It was found that the cis-aconityl bond released DOX much faster than the hydrazone linkage. However, intact DOX was not fully generated from the cis-aconityl bond making the hydrazone linkage favorable because it completely regenerated intact DOX [17]. Therefore, the hydrazone linkage was also examined for long term release and found to deliver the conjugated DOX over a two-week period (Figure 9).
Figure 9. Long-term release of DOX conjugated to micelles via a hydrazone linkage [17].
Figure 8. Short-term release from micelles with DOX conjugated A) via hydrazone linkage and B) via cis-aconityl linkage [17].
The problem with this study was that it was looking solely at PMs for intravenous
delivery. While the release profile exhibited controlled release, the pH favored release at more acidic conditions which would be problematic if these linkages were used for release in the GIT. The importance of these experiments was that it demonstrates chemical conjugation as a method to improve the drug loading and release profiles. However, it would be necessary to find chemical links that are stable at the acidic pH of the stomach until the PM reaches the more basic pH of the intestines, where release is desired by oral DDSs. 3.2 pH-responsive Polymeric Micelles
Acrylic acid monomers have been investigated as a pH-sensitive drug delivery
system because the pKa of its acidic hydrogen is between that of the stomach and
intestines allowing for controlled release in the GIT. Acrylic acid becomes deprotonated, leading to hydrophilicity, when a solution has a pH more acidic than 4.5 [19]. At the pH of the stomach, acrylic acid will be deprotonated physically stabilizing the PM due to shrinkage of the polymer network. However, at the pH of the intestines, acrylic acid will become protonated, leading to swelling of the PM. This swelling allows for release of the drug encapsulated in the hydrophobic core of the PM.
An earlier study demonstrated the pH sensitivity of acrylic acids by looking at 14
different block copolymers that had different acrylates and chain lengths. These block copolymers, of the general form PEG-b-poly(alkyl(meth)acrylate-co-methacrylic acid), were shown to self-assemble into micelles with a high loading capacity and pH-dependent release profile [20]. The drug examined was candesartan cilexetil (CDN) because it displays poor water solubility and bioavailability. Furthermore, the absorption of CDN occurs mainly in the small intestine, so determining a pH-responsive block copolymer that spontaneously forms PMs would be useful for similar drugs that cannot pass the GIT barriers [20]. Results indicated that the hydrophobicity of the micelle core was able to minimize the typical burst phase release of the drug on exposure to acidic pH, but a shift from acidic to neutral pH increased the release rate. The pH-dependence was clearly dependent on the type of acrylate and whether it was ionizable (Figure 10) [20]. This work demonstrated that PMs could be modified to have a controlled release of the encapsulated drug by including a stimulus responsive moiety in the hydrophobic block.
An improvement of this technique was done by introducing a hydrotropic agent
into the hydrophobic core block which increased drug loading capacity and yielded better aqueous stability because of the hydrophobic and hydrotropic interactions in the micellar core. A hydrotropic agent is a low molecular weight compound that can be absorbed by the body following oral delivery and is similar to micelle block copolymers in that it typically contains a hydrophilic and hydrophobic segment but is unable to spontaneously undergo self-aggregation. The experiment investigated the loading and release of paclitaxel (PTX), a poorly water soluble (0.3 μg/mL) anticancer drug, in simulated intestinal and gastric fluids [19]. A screening process found that the polymer N,N-dimethylnicotinamide (DENA) greatly improved the water solubility of PTX to 39 mg/ml [21]. The group modified DENA to include a vinylbezyloxy group so that it could easily be polymerized when incorporated in copolymers of PEG and tert-butyl acyrlate. Acrylic acid contents from 0-50% were studied to determine effects on the release profile. The
introduction of acrylic acid moieties to the hydrotropic polymers resulted in PMs with a pKa of 4.5 and CMC of 80 μg/mL [19]. The hydrotropic PMs exhibited better loading
due to the increase in possible sites for hydrogen-bonding which prevents salting effects that might occur in typical loading of PTX. Furthermore the maximum loading efficiency occurred at conditions that did not disrupt micelle formation [19].
Figure 10. A) In vitro release of CDN from different iso-butyl acrylates containing PM in the presence and absence (open circle) of copolymer. B) Effect of alkyl(meth)acrylate on in vitro release of CDN from PM. C) Effect of tert-butyl methacrylate substitution by methyl methacrylate on in vitro release of CDN from PM. The arrow corresponds to the change in pH from 1.2 to 7.2 [20].
in vitro release of PTX from the PMs was measured using simulated
intestinal fluid (SIF) and simulated gastric fluid (SGF) that mimic the conditions experienced in the body. The PM was first exposed to SGF for one hour, corresponding to typical stomach transit times, followed by an extended exposure to SIF. The cumulative release rates in the two fluids were monitored and compared to determine if the release in stomach conditions are significant (Figure 11). Above 20% acrylic acid content, PTX release in SIF was shown to be effectively increased and demonstrated a significant difference from fluid released in the SGF [19]. The work demonstrated the PTX release rate from the PM was stimulated by the environmental pH and was able to preferentially release in simulated intestinal conditions where delivery of oral drugs is preferred.
Figure 11. Cumulative release profile of PTX from hydrotropic PMs with varying acrylic acid (AA) content in (a) simulated intestinal fluid (SIF) and (b) simulated gastric fluid (SGF). (c) Comparison of amount of PTX release in SGF (0-1hr) and SIF (1-2hr) to determine the difference in rate of release. PTX – paclitaxel without micelle carrier, HP0 – 0% AA, HP1 – 10% AA, HP2 – 20% AA, HP3 – 30% AA, HP4 – 40% AA, HP5 – 50% AA [19].
Current results indicate that PMs can easily be modified to respond to external
conditions that would improve release in the desired area of the GIT. Furthermore, they are able to substantially increase the water solubility of the drug, thus improving bioavailability. Further study needs to focus on the absorption of the drug into the blood stream following this controlled delivery to determine if it still exhibits the desired pharmacological effect. Incorporation of acrylic acid and hydrotropic agents has demonstrated that PMs can be modified to overcome two of the three barriers that make oral drug delivery so difficult. 3.3 Oral vs. Intravenous Administration of Polymeric Micelles
Little work has compared oral delivery to intravenous delivery of poorly soluble
drugs. PM work has also been limited in studying the passage through the intestinal barrier after oral administration. A recent work looked at this ability of a PM made from mono-methyletherpoly(oxyethylene glycol)-poly(caprolactone-co-trimethylene carbonate) (PEG-P(CL-co-TMC)) to transport risperidone across an intestine cell culture line, Caco-2, and the biological fate of the PM once in the blood stream [22]. Results indicated that the PM was able to cross the intestinal membrane when well above the CMC, achieving a bioavailability of 40%, and the PMs were excreted within 24 hours [22]. This long excretion time indicates long acting drug delivery compared to intravenous PMs which were excreted after 8 hours. These results demonstrated that the PM may be orally
available and capable of delivering dose to specific organs over an extended time period. In comparison to intravenous administration, slightly lower percentages of the total administered dose were recovered in the organs orally, but the plasma levels of orally administered drug were similar to intravenously delivered PMs (Figures 12 and 13). The conclusion from this work is that oral delivery by PMs exhibits similar delivery properties to intravenous delivery. The benefit of this is that the more patient-friendly oral delivery method could prove to be a viable drug delivery method when PMs are used, while still giving the pharmacological effects of an intravenous dose.
Figure 13. Plasma profile of
Figure 12. Mean tissue levels of
unchanged risperidone (square) and
radioactively labeled PEG-P(CL-co-
metabolically formed 9-
TCM) as % of total dose delivered to
hydroxyrisperidone (triangle) after
the liver (circle), kidney (square), and
single oral (empty symbols) and
spleen (triangle) after (A) oral and (B)
intravenous (filled symbols)
intravenous administration [22].
administration [22].
4. Conclusion
Polymeric micelles exhibit many properties suitable for delivery vehicles of
biologic drugs. PMs are small, on the order of 10 nm, and can easily be surface modified to improve uptake via the GIT during oral delivery. Current work has been lacking on examining the uptake once in the GIT and has instead focused on modifying the PMs to respond to external stimuli that increase the drug incorporation and release in the intestines. In order to determine if PMs can be clinically relevant, dosing needs to be analyzed to make sure that upon release the drug is indeed being transported to the bloodstream and having the desired pharmacological effect.
While the oral route is the easiest and preferred method of administration, PMs
can also be administered in other ways, such as intravenously, indicating its potential of a future delivery vehicle for multiple types of medications. The difficulties posed by intravenous delivery are different from those presented here, but the ability to modify the PM surface for targeting specific cells makes it a viable option [5]. Also, the nanometer size allows PMs to avoid many of the excretion pathways making it possible to accumulate at the targeted site, improving the efficacy of the treatment [7]. These multiple pathways, if found to lead to efficient and effective delivery, could make PMs the future of medical delivery systems for biologics.
References
[1] FDA Data Standards Manual C-DRG-00301. Route of Administration. Center for
Drug Evaluation and Research. 2006. http://www.fda.gov/cder/dsm/DRG/drg00301.htm
[2] Colombo P, Sonvico F, Colombo G, Bettini R. Novel Platforms for Oral Drug
Delivery. Pharmaceutical Research 2009.
[3] Morishita Mariko, Peppas Nicholas A. Is the oral route possible for peptide and
protein drug delivery? Drug Discovery Today 2006; 11:905-910.
[4] Lavelle EC. Targeted Delivery of Drugs to the Gastrointestinal Tract. Critical Revies
in Therapeutic Drug Carrier Systems 2001; 18(4):341-386.
[5] Chen Hongming, Langer Robert. Oral particulate delivery: status and future trends.
Advanced Drug Delivery Reviews 1998; 34:339-350.
[6] Jarvis LM. Hope in a Pill. Chemical and Engineering News 2009; 87(114):10-15. [7] Torchilin VP. Micellar Nanocarriers: Pharmaceutical Perspectives. Pharmaceutical
Research. 2007; 24(1):1-16.
[8] http://www.justlaparoscopy.com/images/gastrointestinal_tract.jpg [9] Francis Mira F, Cristea Mariana, Winnik Françoise M. Polymeric micelles for oral
drug delivery: Why and how. Pure Applied Chemistry 2004; 76:1321-1335.
[10] Mondon Karine, Gurny Robert, Möller Michael. Colloidal Drug Delivery Systems –
Recent Advances with Polymeric Micelles. Chimia 2008; 62:832-840.
[11] Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of
drugs in the gastrointestinal tract. Advanced Drug Delivery Reviews 2001; 46:75-87.
[12] Bromberg, Lev. Polymeric micelles in oral chemotherapy. Journal of Controlled
Release 2008; 128:99-112.
[13] Torchilin Vladimir P. Structure and design of polymeric surfactant-based drug
delivery systems. Journal of Controlled Release 2001; 73:137-172.
[14] Romani Ana Paula, Machado Antonio Eduardo da Hora, Hioka, Noboru, Severino
Divinomar, Baptista Mauricio S, Codognoto Lúcio, Rodrigues Maira R, Moisés de Oliveira Hueder Paulo. Spectrofluorimetric Determination of Second Critical Micellar Concentration of SDS and SDS/Brij 30 Systems. J Fluoresc 2009; 19:327-332.
[15] Aliabadi HM, Lavasanifar A. Polymeric micelles for drug delivery. Expert Opinion
on Drug Delivery 2006; 3(1):139-162.
[16] Bilgiçer B. Biomedical Engineering. Class Notes Biomolecular Topics in
Engineering. March 24, 2009.
[17] Yoo HS, Lee EA, Park TG. Doxorubicin-conjugated biodegradable polymeric
micelles having acid-cleavable linkages. Journal of Controlled Release 2002; 82:17-27.
[18] Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin
conjugated PLGA-PEG block copolymer. Journal of Controlled Release 2001; 70:63-70.
[19] Kim S, Kim JY, Huh KM, Acharya G, Park K. Hydrotropic polymer micelles
containing acrylic acid moieties for oral delivery of paclitaxel. Journal of Controlled Release 2008; 132:222-229.
[20] Satturwar P, Eddine MN, Ravenelle F, Leroux JC. pH-responsive polymeric
micelles of poly(ethylene glycol)-b-poly(alkyl(meth)acrylate-co-methacrylic acid): Influence of the copolymer composition on self assembling properties and release of candesartan cilexetil. European Journal of Pharmaceutics and Biopharmaceutics 2007; 65:379-387.
[21] Lee J, Lee SC, Acharya G, Chang CJ, Park K. Hydrotropic solubilization of
paclitaxel: analysis of chemical structures for hydrotropic property. Pharmaceutical Research 2003; 20(7):1022-1030.
[22] Mathot F, van Beijsterveldt L, Préat V, Brewster M, Ariën A. Intestinal uptake and
biodistribution of novel polymeric micelles after oral administration. Journal of Controlled Release 2006; 111:47-55.
Source: https://ame.nd.edu/research/faculty-research-labs/rroeder/classes/ame-50571/term-paper/polymer-micelles-the-future-of-oral-drug-delivery.pdf
Pii: s0168-8510(00)00066-x
Health Policy 52 (2000) 129 – 145 The cost of prescription medicines to patients Peter R. Noyce a,*, Christine Huttin b, Vicenzo Atella c, Gerhard Brenner d, Flora M. Haaijer-Ruskamp e, Maj-Britt Hedvall f, Reli Mechtler g a School of Pharmacy and Pharmaceutical Sciences, Uni6ersity of Manchester, Oxford Road, Manchester, M13 9PL, UK
Gaafar_f_rev
STIMULATION AND CONTROL OF E. COLI BY USING AN EXTREMELY LOW FREQUENCY MAGNETIC FIELD EL-SAYED A. GAAFAR*, MAGDA S. HANAFY**, EMAN Y. TOHAMY***, MONA H. IBRAHIM** * Biophysics Department, Faculty of Science, Cairo University, Egypt ** Physics Department, Faculty of Science, Zagazig University, Egypt *** Botany Department, Faculty of Science, Zagazig University, Egypt