Rappel physiologique
N° d'ordre : 209-2009 Présentée devant L'UNIVERSITE CLAUDE BERNARD - LYON 1 Pour l'obtention du DIPLOME DE DOCTORAT (arrêté du 7 août 2006) Présentée et soutenue publiquement le 18 novembre 2009 M. Burhan KNOUZY Chloroacétaldéhyde : de l'implication dans les mécanismes physiopathologiques de la néphrotoxicité de l'ifosfamide à la contribution à son effet anticancéreux Directeur de thèse : Pr. Gabriel BAVEREL Pr. Jean-Claude DUSSAULE Dr. Xavier COUMOUL Pr. Gabriel BAVEREL Dr. Christian MICHOUDET UNIVERSITE CLAUDE BERNARD - LYON 1 Président de l'UniversitéM. le Professeur L. Collet
Vice-président du Conseil Scientifique M. le Professeur J-F. Mornex Vice-président du Conseil d'Administration M. le Professeur G. Annat Vice-président du Conseil des Etudes et de la Vie Universitaire M. le Professeur D. Simon Secrétaire Général COMPOSANTES SANTE Faculté de Médecine Lyon Est – Claude Bernard Directeur : M. le Professeur J. Etienne Faculté de Médecine Lyon Sud – Charles Mérieux Directeur : M. le Professeur F-N. Gilly UFR d'Odontologie Directeur : M. le Professeur D. Bourgeois Institut des Sciences Pharmaceutiques et Biologiques Directeur : M. le Professeur F. Locher Institut des Sciences et Techniques de Réadaptation Directeur : M. le Professeur Y. Matillon Département de Formation et Centre de Recherche en Biologie Directeur : M. le Professeur P. Farge COMPOSANTES SCIENCES ET TECHNOLOGIE Faculté des Sciences et Technologies Directeur : M. Le Professeur F. Gieres UFR Sciences et Techniques des Activités Physiques et Sportives Directeur : M. C. Collignon Observatoire de Lyon Directeur : M. B. Guiderdoni Institut des Sciences et des Techniques de l'Ingénieur de Lyon Directeur : M. le Professeur J. Lieto Institut Universitaire de Technologie A Directeur : M. le Professeur C. Coulet Institut Universitaire de Technologie B Directeur : M. le Professeur R. Lamartine Institut de Science Financière et d'Assurance Directeur : M. le Professeur J-C. Augros Institut Universitaire de Formation des Maîtres Directeur : M R. Bernard TABLE DES MATIERES Aldéhyde déshydrogénase Chloroacétaldéhyde Cyclophosphamide Dihydroxyacetone phosphate 5,5'-Dithio-bis-2-nitrobenzoic acide F1,6-Bpase: Fructose 1,6-bisphosphatase Fructose 1,6-bisphosphate Fructose 2,6-bisphosphate Fructose 6-phosphate Glucose 6-phosphatase Glucose 6-phosphate Glucose 6-phosphate déshydrogénase Glyceraldéhyde 3-phosphate Glyceraldéhyde 3-phosphate déshydrogénase 4-Hydroxy ifosfamide Lactate déshydrogénase Phosphoénolpyruvate Phosphoénolpyruvate carboxykinase Phosphofructokinase 3-Phosphoglycérate Phosphoglycérate Kinase Pyruvate carboxylase Pyruvate déshydrogénase Résumé en français Le chloroacétaldéhyde (CAA), un des principaux produits du métabolisme hépatique de l'ifosfamide (IFO), est considéré comme responsable de la néphrotoxicité de ce médicament. Les mécanismes exacts de cette néphrotoxicité ne sont pas complètement élucidés. Dans la première partie de cette étude, nous avons essayé de préciser les mécanismes physiopathologiques de la toxicité du CAA sur un modèle de tranches de cortex rénal de rat, puis, dans la deuxième partie, nous avons recherché un effet anticancéreux éventuel du CAA sur des cellules de cancer du sein humain (MCF-7). La néphrotoxicité du CAA, utilisé à des concentrations proches de celles mesurées chez les patients traités par l'IFO, soit 0 - 75 µM, s'est manifestée par une chute d'ATP, du glutathion et des thiols cellulaires ainsi que par une inhibition du métabolisme du lactate (inhibition de la consommation du lactate et de la néoglucogenèse). Certaines enzymes de la néoglucogenèse, notamment la glyceraldéhyde 3-phosphate déshydrogénase, ont été inhibées par le CAA. Le complexe I de la chaîne respiratoire mitochondriale ainsi que l'oxydation de [U-14C]- lactate ont été également inhibées par le toxique. D'autre part, le CAA (à 10 et 25 µM) a inhibé la prolifération des cellules MCF-7 sans que cette inhibition soit accompagnée d'une chute d'ATP cellulaire. Le transport cellulaire et le métabolisme du glucose ainsi que certaines enzymes de la glycolyse ont été également inhibés par le CAA. Parmi celles-ci, l'hexokinase semble être l'enzyme qui catalyse l'étape limitante de la voie de la glycolyse. En conclusion, le CAA est bien impliqué dans les mécanismes de la néphrotoxicité de l'IFO, mais de plus, il pourrait, via l'inhibition de la glycolyse, contribuer à l'effet thérapeutique de l'IFO. Résumé en anglais Chloroacetaldehyde (CAA), one of the main products of ifosfamide (IFO) hepatic metabolism, is considered as responsible of IFO nephrotoxicity. The mechanisms of this nephrotoxicity are not completely known. In the first part of this study, we tried to clarify the pathophysiological mechanisms of CAA toxicity using precision-cut rat renal cortical slices, then, in the second part, we looked for a possible anticancerous effect of CAA on human breast cancer cells (MCF-7). Using clinically-relevant concentrations (0-75 µM), CAA nephrotoxicity was demonstrated by the depletion of ATP and glutathione and by the inhibition of lactate metabolism (inhibition of lactate consumption and gluconeogenesis). Some of the gluconeogenic enzymes, mainly glyceraldehyde 3-phosphate dehydrogenase, were inhibited by CAA. The complex I of the mitochondrial respiratory chain as well as [U-14C]-lactate oxidation were also inhibited by CAA. On the other hand, CAA (10 and 25 µM) inhibited MCF-7 cell proliferation which was not accompanied by cellular ATP depletion. Glucose transport and metabolism as well as some of the glycolytic enzymes were also inhibited by CAA. Hexokinase seems to be the rate-limiting enzyme of glycolysis. In conclusion, CAA is implied in the mechanisms of IFO-induced nephrotoxicity; furthermore, it could, via the inhibition of the glycolytic pathway, contribute to the therapeutic effect of IFO. L'ifosfamide (IFO), médicament anticancéreux, est un agent alkylant de la famille des oxazaphosphorines. Depuis son introduction en clinique au début des années 1960, il a été utilisé dans de nombreux protocoles antinéoplasiques, mais son utilisation était limitée à cause de ses effets secondaires. L'IFO est un promédicament. Il est métabolisé dans le foie par le cytochrome P450 pour donner la moutarde azotée, la molécule active. Son métabolisme conduit à la production du chloroacétaldéhyde (CAA) et d'acroléine. La toxicité urothéliale due à l'acroléine a longtemps empêché l'administration d'IFO à des doses suffisantes pour être efficaces sur les tissus cancéreux, mais depuis l'introduction du mesna (2-mercaptoéthanesulfonate de sodium), cette toxicité a pu être contrôlée. La néphrotoxicité de l'IFO, notamment tubulaire proximale, reste jusqu'à présent l'effet secondaire le plus important lors du traitement par l'IFO. Bien que le CAA soit considéré par la plupart des auteurs comme étant le responsable de la néphrotoxicité de l'IFO, les mécanismes physiopathologiques et les lieux de son action ne sont pas complètement connus. Il est important de préciser ces mécanismes afin de contrôler la toxicité de l'IFO. Il a été montré dans une étude préalablement réalisée dans notre laboratoire que le CAA exerce des effets toxiques sur les tubules proximaux isolés du rein humain. La toxicité du CAA s'est manifestée par une diminution d'ATP, de glutathion et des thiols intracellulaires, ainsi que par une inhibition du métabolisme du lactate et de la néoglucogenèse. Cette toxicité avait été obtenue en utilisant des concentrations très élevées de CAA (> 500 µM). Notre étude a deux objectifs. Nous allons, dans la première partie, montrer l'effet toxique du CAA sur le tissu rénal et préciser les mécanismes sous-jacents de la néphrotoxicité induite par ce métabolite. Dans la deuxième partie, nous allons rechercher un effet toxique éventuel du CAA sur des cellules de cancer du sein humain (MCF-7), un effet qui pourrait contribuer à l'efficacité thérapeutique de l'IFO. Nous avons voulu étudier l'effet néphrotoxique du CAA en appliquant des concentrations faibles du toxique, proches de celles mesurées dans le sang et les urines des patients traités par l'IFO. Pour pouvoir réaliser des incubations d'assez longue durée (24 heures), nous avons utilisé un modèle de tranches tissulaires. Ce modèle, qui jouit d'un regain d'intérêt dans les études pharmacologiques et toxicologiques, permet de réaliser des expériences in vitro pour étudier le métabolisme et le transport cellulaire pendant des périodes longues (2-3 jours) contrairement aux tubules isolés, dont l'incubation ne peut pas durer plus de quelques heures. Dans la première partie, nous avons étudié l'effet toxique du CAA sur le tissu rénal en utilisant des tranches fines et calibrées de cortex rénal de rat. Les concentrations appliquées du CAA variaient entre 0 et 75 µM. Le métabolisme du lactate (à une concentration proche de la concentration physiologique, soit 1 mM) est étudié en présence ou en absence du CAA. La consommation du lactate, la néoglucogenèse et l'accumulation du pyruvate, ainsi que les paramètres de la viabilité cellulaire (l'ATP et le glutathion) ont été mesurés. Nous avons également étudié l'activité de certaines enzymes de la néoglucogenèse, la respiration mitochondriale et l'oxydation du lactate marqué ([U-14C]-lactate). Les résultats de cette première partie montrent clairement l'effet toxique du CAA sur le tissu rénal. Cet effet se manifeste par une chute du glutathion et d'ATP et par une inhibition de la néoglucogenèse. Certaines enzymes de cette voie métabolique, notamment la glyceraldéhyde 3-phosphate déshydrogénase (GAPDH), sont inhibées. La chaîne respiratoire des mitochondries isolées des tranches de cortex rénal ainsi que l'oxydation de [U-14C]-lactate sont également inhibées par le CAA. Dans la deuxième partie, nous nous sommes posés la question de savoir si le CAA pouvait, en inhibant la GAPDH (qui est à la fois une enzyme de la néoglucogenèse et de la glycolyse), inhiber la voie de la glycolyse et, par conséquent, altérer le métabolisme et la croissance des cellules cancéreuses. Les cellules cancéreuses possèdent une capacité glycolytique très élevée même en présence d'oxygène (effet Warburg). Elles comptent sur la glycolyse comme source principale pour la production d'énergie et des précurseurs des biosynthèses. L'inhibition de cette voie métabolique pourrait avoir des effets négatifs sur le métabolisme et la survie des cellules cancéreuses, conduisant à la mort cellulaire. En s'appuyant sur l'effet Warburg, l'inhibition de la glycolyse est proposée par de nombreux auteurs comme une stratégie pour tuer sélectivement les cellules cancéreuses. Nous avons donc recherché les effets toxiques éventuels du CAA sur des cellules de cancer du sein humain, MCF-7. L'utilisation de ces cellules dans les études de cancer est bien connue, et elle a été rapportée dans de nombreuses publications. L'incubation des cellules est réalisée en présence de concentrations variables de CAA (0 - 50 µM). La prolifération des cellules, l'énergie cellulaire, le métabolisme et le transport du glucose ainsi que l'activité de certaines enzymes de la glycolyse ont été étudiés dans des conditions de normoxie puis dans des conditions d'hypoxie (5% O2), qui est une caractéristique importante de l'environnement tumoral. Les résultats de cette deuxième partie montrent que le CAA, à la plus forte concentration (50 µM), entraîne une destruction des cellules et, qu'à plus faible concentration, il inhibe le métabolisme et le transport du glucose dans les cellules MCF-7. Il inhibe également certaines enzymes de la glycolyse, notamment l'hexokinase (HK). Contrairement à ce qui était prévu, le CAA n'a pas provoqué une chute de l'ATP cellulaire, même dans les conditions d'hypoxie. Cette dernière n'était probablement pas suffisamment sévère pour induire une telle inhibition dans nos Nos résultats montrent, malgré tout, que le CAA exerce un effet toxique sur les cellules MCF-7. Il peut donc contribuer à l'effet thérapeutique de l'IFO, expliquant ainsi l'efficacité thérapeutique supérieure de l'IFO par rapport à son isomère structurel le cyclophosphamide (CP), dont le métabolisme produit des quantités moins importantes de CAA.
II- RAPPELS ET DONNEES BIBLIOGRAPHIQUES
1. RAPPEL PHYSIOLOGIQUE
1-1. PHYSIOLOGIE RENALE
Les reins, chez l'homme, sont situés dans les fosses lombaires entre la onzième
vertèbre dorsale et la troisième vertèbre lombaire. Ils pèsent 120-150 grammes et
mesurent environ 12 cm de longueur, 6 cm de largeur et 3 cm d'épaisseur. En coupe
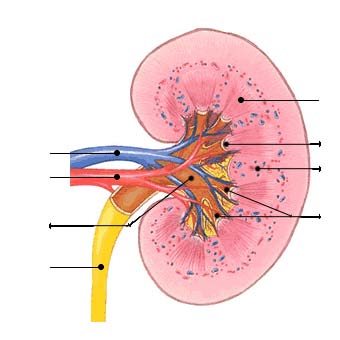
frontale (Figure 1), on distingue de l'extérieur à l'intérieur : la capsule fibreuse, le
cortex, la médullaire et le bassinet. Le système urinaire est complété par l'uretère et
Figure 1 : Anatomie rénale, coupe longitudinale
Les reins sont irrigués par 25% du débit sanguin cardiaque (1200 ml/minute). Le
sang entre dans le rein par l'artère rénale et chemine à travers un réseau d'artères et
d'artérioles jusqu'à un glomérule. De chaque glomérule part une artériole efférente
qui se ramifie en de nombreux capillaires péritubulaires qui vont se jeter dans les
veinules rénales. Le réseau veineux est identique au réseau artériel et se termine au
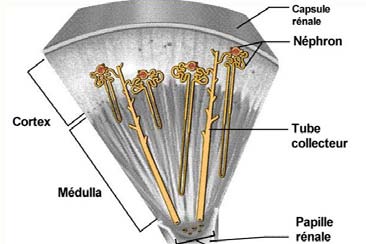
niveau du hile par la veine rénale. Chaque rein est composé d'environ un million de
tubules fins appelés les néphrons (Figure 2). Il existe deux types de néphrons, les
néphrons corticaux superficiels et les néphrons juxtamédullaires qui diffèrent par la
localisation du glomérule dans la zone corticale et la longueur de l'anse de Henlé.
Figure 2 : Anatomie du néphron
Tubule contourné distal
Tubule contourné
La partie initiale de chaque néphron est formée par le glomérule de Malpighi qui est
entouré de la capsule de Bowman. Se succèdent ensuite le tubule contourné
proximal, l'anse de Henlé, le tubule contourné distal et les tubes collecteurs qui se
jettent dans le bassinet en traversant les papilles rénales. Le glomérule permet la
filtration du sang, le tubule rénal reçoit ce filtrat et le transforme en urine qui est
déversée dans le bassinet de chaque rein.
Les rôles physiologiques des reins sont multiples [1]. Ils interviennent dans
l'excrétion des déchets métaboliques terminaux (urée, créatinine, acide urique), dans
la détoxication et l'élimination des produits exogènes (toxiques, médicaments) et
dans le contrôle de l'équilibre acido-basique. Les reins maintiennent la composition
du milieu intérieur en équilibrant les entrées et les sorties d'eau et d'électrolytes
(sodium, potassium, chlorure). Ils participent également à la conversion de la
vitamine D en son métabolite actif, à la synthèse et la sécrétion d'érythropoïétine, à
la régulation de la pression artérielle (synthèse de la rénine), et au catabolisme
d'hormones polypeptidiques (insuline, glucagon) et de facteurs de croissance
(insuline growth factor-1). Enfin, ils interviennent dans le catabolisme des protéines
de bas poids moléculaire.
Plusieurs paramètres contribuent à la grande sensibilité du rein vis-à-vis de
substances exogènes. Le rein présente un fort débit sanguin et il a un rôle de
concentration et d'élimination des déchets via l'urine. Le cortex possède un rapport
flux sanguin/masse de tissu élevé. Il reçoit environ 90% du flux sanguin rénal et il est
le plus exposé aux néphrotoxiques. Le tubule contourné proximal, qui forme la
majorité de la masse du cortex, est donc la cible principale des toxiques rénaux ; de
ce fait, il possède un équipement enzymatique très riche (cytochrome P450,
cofacteurs, enzymes du métabolisme du glutathion, de la néoglucogenèse) [2] et de
nombreux transporteurs (glucose, lactate, intermédiaires de cycle du Krebs, acides
aminés). De plus, il a été montré que la production d'ions ammonium à partir de la
glutamine (via la glutaminase) se situait au niveau du tubule proximal [3] et que le
lactate était un des principaux substrats oxydés par le tubule proximal [4].
1-2. LA NEOGLUCOGENESE ET LA GLYCOLYSE
1-2-1. La néoglucogenèse
La voie de la néoglucogenèse [5] permet la synthèse du glucose à partir des
précurseurs non glucidiques (Figure 3). La néoglucogenèse est importante pour
assurer les besoins en glucose au cours d'une période longue de jeûne ou pendant
des périodes d'exercice intense, ou quand les réserves directes de glucose ne sont
pas suffisantes.
Les précurseurs non glycolytiques de glucose entrent dans cette voie principalement
sous forme de pyruvate, d'oxaloacétate, et de dihydroxyacétone phosphate (DHAP).
Figure 3 : Schéma de la néoglucogenèse
Glucose 6-phosphatase
Glucose 6-phosphate
Phosphoglucose isomérase
Fructose 6-phosphate
Fructose 1,6-bis phosphatase
Fructose 1,6-bisphosphate
Dihydroxyacétone phosphate
Glycéraldéhyde 3-phosphate
Glycéraldéhyde 3-phosphate déshydrogénasease
Certains acides aminés
Phosphoglycérate
Lactate déshydrogénase
3-Phosphoglycérate
Pyruvate
2-Phosphoglycérate
Pyruvate carboxylase
Phosphoénolpyruvate
Phosphoénolpyruvate
Cycle des acides
tricarboxyliques
α-cétoglutarate
Certains acides aminés
Les précurseurs non glucidiques principaux sont le lactate, les acides aminés et le
glycérol. Le lactate est formé par le muscle squelettique au cours de l'activité quand
la vitesse de la glycolyse excède la vitesse métabolique du cycle des acides
tricarboxyliques et la chaîne respiratoire. Les acides aminés proviennent des
protéines de l'alimentation et de la destruction des protéines du muscle squelettique
au cours du jeûne. L'hydrolyse des triacylglycérols dans les cellules adipeuses
fournit du glycérol et des acides gras. Plusieurs des réactions qui transforment le
pyruvate en glucose sont communes à la voie de la glycolyse. La néoglucogenèse,
cependant, a besoin de quatre nouvelles réactions pour court-circuiter les réactions
essentiellement irréversibles correspondantes de la glycolyse. Du pyruvate est
carboxylé en oxaloacétate dans les mitochondries. L'oxaloacétate est décarboxylé et
phosphorylé dans le cytosol en phosphoénolpyruvate (PEP). Deux liaisons
phosphate riches en énergie sont consommées dans ces réactions catalysées par la
pyruvate carboxylase (PC) et la phosphoénolpyruvate carboxykinase (PEPCK). Les
autres réactions distinctives à la néoglucogenèse sont les hydrolyses du fructose
1,6-bisphosphate (F1,6-P2) et du glucose 6-phosphate (G6-P) qui sont catalysées
par des phosphatases spécifiques (voir ci-dessous).
Le site principal de la néoglucogenèse est le foie. Elle se produit également dans le
cortex rénal, mais la quantité totale de glucose formée n'est qu'environ un dixième
de celle formée dans le foie, en raison de la plus petite taille des reins. La
néoglucogenèse dans le foie et le rein aide au maintien de la concentration de
glucose dans le sang, de telle sorte que le cerveau et le muscle puissent extraire
suffisamment de glucose pour leurs besoins métaboliques.
Le rein est composé de deux parties physiologiquement différentes en ce qui
concerne le métabolisme du glucose. L'utilisation du glucose est prédominante au
niveau de la médullaire alors que la néoglucogenèse prédomine dans le cortex rénal
[2]. Cette répartition fonctionnelle résulte de différences de distribution des enzymes
le long du néphron. Les cellules de la médullaire possèdent une activité enzymatique
importante de la glycolyse et de la phosphorylation du glucose. Elles ne possèdent
pas de glucose 6-phosphatase (G6-Pase) et d'autres enzymes de la
néoglucogenèse. En revanche, les cellules du cortex rénal possèdent les enzymes
de la néoglucogenèse, et donc, peuvent fabriquer du glucose. Autrement dit, tout le
glucose produit par un rein normal vient exclusivement de la néoglucogenèse du
cortex et toute l'utilisation rénale du glucose a lieu dans la médullaire. Le lactate, la
glutamine, l'alanine et le glycérol représentent les précurseurs principaux de la
néoglucogenèse rénale (> 90%).
1-2-2. La glycolyse
La glycolyse [6] est un ensemble de réactions qui transforme le glucose en pyruvate
(Figure 4). Dans les organismes aérobies, la glycolyse est le prélude du cycle des
acides tricarboxyliques et de la chaîne de transport d'électrons où la plus grande
partie d'énergie libre du glucose est récupérée. Les 10 réactions de la glycolyse
s'effectueront dans le cytosol. Dans les premières étapes, le glucose est transformé
en F1,6-P2 par une phosphorylation, une isomérisation et une seconde réaction de
phosphorylation. Deux molécules d'ATP sont consommées par une molécule de
glucose dans ces réactions. Dans un deuxième temps, le F1,6-P2 est clivé par
l'aldolase en DHAP et glycéraldéhyde 3-phosphate (GAP) qui sont facilement inter-
convertibles. Le GAP est alors oxydé et phosphorylé pour former le 1,3-
bisphosphoglycérate, un acyl phosphate qui possède un potentiel élevé de transfert
de phosphate. Le 3-phosphoglycérate (3-PG) est alors formé et en même temps un
ATP est produit. Dans la dernière étape de la glycolyse, le PEP, un second
intermédiaire avec un potentiel élevé de transfert de phosphate, est formé par un
déplacement de phosphoryle et une déshydratation. Un autre ATP est formé lorsque
le PEP est transformé en pyruvate. Il existe un gain net de deux molécules d'ATP au
cours de la formation de deux molécules de pyruvate à partir d'une seule molécule
L'accepteur d'électron dans l'oxydation du GAP est le NAD+ qui doit être régénéré
pour que la glycolyse puisse se poursuivre. Chez les organismes aérobies, le NADH
formé au cours de la glycolyse transfère ses électrons à l'O2 à travers la chaîne
respiratoire, ce qui permet la régénération du NAD+. Dans des conditions anaérobies,
le NAD+ est régénéré par la réduction du pyruvate en lactate. Chez certains micro-
organismes, le NAD+ est régénéré par la synthèse de lactate ou d'éthanol à partir du
pyruvate. Ces deux mécanismes sont des fermentations.
Figure 4 : La voie de la glycolyse et des pentoses phosphate. Les trois réactions
irréversibles sont catalysées par l'hexokinase, la phosphofructokinase et la pyruvate kinase
Glucose 6-phosphate
Gluconolactone 6 phosphate
déshydrogénase
Glucose 6-phosphate
6 phosphogluconate
Fructose 6-phosphate
Phosphofructokinase
Dihydroxyacétone phosphate
Fructose 1,6-bisphosphate
Ribose 5-phosphate
Glycéraldéhyde 3-phosphate
Glycéraldéhyde 3-phosphate
déshydrogénasease
Phosphoglycérate
3-Phosphoglycérate
2-Phosphoglycérate
Phosphoénolpyruvate
Pyruvate
La voie glycolytique possède un double rôle : elle dégrade le glucose pour former
l'ATP et elle fournit des éléments pour les réactions de biosynthèse des
macromolécules. La vitesse de conversion du glucose en pyruvate est régulée pour
assurer ces deux importants besoins cellulaires.
Dans les conditions physiologiques, les réactions de la glycolyse sont réversibles,
excepté pour celles catalysées par l'hexokinase (HK), la phosphofructokinase (PFK)
et la pyruvate kinase (PK). La PFK, le plus important élément de contrôle de la
glycolyse, est inhibée par des concentrations élevées d'ATP et de citrate, et est
activée par l'AMP et le fructose 2,6-bisphosphate (F2,6-P2). Dans le foie, ce dernier
signale que le glucose est abondant. La PFK est donc active quand un besoin
survient, soit en énergie, soit en éléments de construction. L'HK est inhibée par le
G6-P qui s'accumule quand la PFK est inactive. La PK, l'autre site de contrôle, est
allostériquement inhibée par l'ATP et l'alanine et est activée par le F1,6-P2. Elle est
pleinement active quand la charge énergétique est faible et quand les intermédiaires
glycolytiques s'accumulent. La PK est régulée par une phosphorylation réversible.
Une concentration faible du glucose dans le sang conduit à la phosphorylation, et
donc la désactivation, de la PK hépatique, diminuant ainsi la consommation du
1-2-3. Les enzymes clés de la néoglucogenèse et la glycolyse
La néoglucogenèse n'est pas simplement l'inverse de la glycolyse. Dans la glycolyse
il y a trois étapes réactionnelles irréversibles, catalysées par l'HK, la PFK et la PK
(Figure 4). Dans la néoglucogenèse, ces réactions sont court-circuitées par les
étapes suivantes (Figure 3).
1- Le PEP est formé à partir du pyruvate par l'intermédiaire de l'oxaloacétate. Le
pyruvate est d'abord carboxylé en oxaloacétate aux dépens de l'ATP. L'oxaloacétate
est ensuite décarboxylé et phosphorylé pour fournir le PEP aux dépens d'une
seconde liaison riche en énergie. La première réaction est catalysée par la PC et la
seconde par la PEPCK.
2- Le fructose-6-phosphate (F6-P) est formé à partir du F1,6P2 par hydrolyse de
l'ester phosphate en C-1. La fructose 1,6-bisphosphatase (F1,6-BPase) catalyse
cette hydrolyse.
3- Le glucose est formé par l'hydrolyse du G6-P, par une réaction catalysée par la
Le G6-P est véhiculé par un transporteur protéique spécifique du cytosol à la lumière
du réticulum endoplasmique où il est hydrolysé par la G6-Pase liée à la membrane.
Le glucose et le Pi reviennent vers le cytosol. La G6-Pase n'est pas présente dans le
cerveau et le muscle ; et donc, le glucose ne peut pas être formé par ces organes.
Six liaisons phosphate riches en énergie sont utilisées pour synthétiser le glucose à
partir du pyruvate alors que seulement deux molécules d'ATP sont formées dans la
glycolyse pour la conversion du glucose en pyruvate.
1-2-4. Régulation de la néoglucogenèse et la glycolyse
La glycolyse et la néoglucogenèse sont réciproquement coordonnées de telle sorte
qu'une voie est relativement inactive alors que l'autre est pleinement active. Si les
deux voies étaient fortement actives en même temps, le résultat ne serait que
l'hydrolyse de quatre liaisons phosphate par cycle réactionnel. La vitesse de la
glycolyse est déterminée par la concentration du glucose, et celle de la
néoglucogenèse par la concentration du lactate et d'autres précurseurs du glucose.
La PFK et la F1,6-BPase sont des points de contrôle importants. Le F2,6-P2, est une
molécule de signalisation intracellulaire provenant du F6-P, et présente à haute
concentration quand le glucose est abondant. Il active la glycolyse et inhibe la
néoglucogenèse en activant la PFK et en inhibant la F1,6-BPase. Au cours du jeûne,
la concentration de F2,6-P2 baisse, ce qui diminue l'activité de la PFK et augmente
celle de la F1,6-BPase. Par conséquent, le F1,6-P2 est transformé en F6-P pour
former du glucose. L'AMP stimule la PFK et inhibe la F1,6-BPase. L'ATP et le citrate
exercent un effet opposé.
La PK et la PC sont régulées par d'autres effecteurs de telle sorte que toutes deux
ne soient pas activées à leur maximum en même temps. Le F1,6-P2 stimule et l'ATP
inhibe la PK, alors que l'acétyl-CoA stimule et l'ADP inhibe la PC.
2-1. PRESENTATION
Les oxazaphosphorines représentent un groupe important d'agents ayant une
activité antinéoplasique et immuno modulatrice. Le CP et l'IFO sont les agents les
plus connus et les plus utilisés de ce groupe. Parmi les membres de cette famille
existent aussi : le trofosfamide, le mafosfamide, le glufosfamide, le bromofosfamide,
etc. Le CP a montré ses effets antinéoplasiques pendant les années 1940 [7] et il a
été introduit dans les protocoles de chimiothérapie. L'IFO, à son tour, est entré en
clinique en fin des années 1960 [8-10] et depuis son introduction il est utilisé dans de
nombreux protocoles de chimiothérapie chez les adultes ainsi que chez les enfants.
2-2. MECANISME D'ACTION
L'effet cytotoxique des oxazaphosphorines dépend de leur capacité d'alkylation
d'ADN. L'activation de l'IFO dans le foie par le cytochrome oxydase P450 mène à la
formation de la moutarde azotée, le métabolite actif de l'IFO. Cette dernière pénètre
dans le noyau (via une diffusion passive et/ou via un transporteur) où elle réagit avec
les bases d'ADN, notamment au niveau de l'azote N-7 de la guanine, en liant leur
groupe actif (le chloroéthyl) à l'azote ce qui aboutit à la formation des ponts inter ou
intra caténaires provoquant une toxicité tissulaire et la mort des cellules [11-13].
D'autres bases d'ADN pourraient être les cibles d'action de la moutarde azotée
comme l'azote N-1 et N-3 de l'adénine, l'azote N-3 de la cytosine, et l'oxygène O-6
de la guanine mais elles sont moins importantes [11].
L'ifosforamide moutarde est un agent alkylant bifonctionnel qui possède deux
groupes chloroéthyl, l'un sur l'azote exocyclique et l'autre sur l'azote endocyclique de
l'oxazaphosphorine. Cela augmente sa capacité à créer des ponts entre les brins
d'ADN (soit au sein du même brin soit entre deux brins opposés) et, par conséquent,
son effet cytotoxique. La formation des ponts mène à la mort cellulaire par apoptose
provoquée par les lésions de l'ADN. Cette apoptose est catalysée par l'activation de
la cascade des caspases [13].
En effet, l'alkylation interfère avec la synthèse et la fonction normale de l'ADN
conduisant à une dépurination et à des erreurs d'appariement des bases ainsi qu'une
inhibition de la séparation et de la duplication d'ADN bicaténaire. Par conséquent,
cela conduit à l'inhibition des mitoses et à la constitution des cellules non viables.
La résistance acquise de la chimiothérapie par les oxazaphosphorines est bien
connue notamment lors du traitement des tumeurs solides [11, 13]. Les origines
biochimiques de cette résistance ne sont pas complètement connues. Plusieurs
facteurs associés à leur métabolisme, à leur transport et à la réponse cellulaire aux
lésions d'ADN provoquées par les oxazaphosphorines peuvent être responsables de
cette résistance [13] (Figure 5).
Figure 5 : Les mécanismes proposés pour expliquer la résistance d'oxazaphosphorines.
D'après Zhang et al., Drug Metabolism and Reviews, 37: 611–703, 2005
Parmi les mécanismes proposés : la diminution de l'activation par les cytochromes
oxydases, l'inhibition de l'entrée ou l'augmentation de la sortie des agents dans et
hors des cellules, l'augmentation de la désactivation des agents dans les cellules,
l'augmentation du contenu intracellulaire des thiols et du glutathion, l'augmentation
de l'activité de la glutathion S transférase, l'augmentation de l'activité aldéhyde
déshydrogénase (ALDH), et l'amélioration de la capacité cellulaire à réparer les
lésions d'ADN [11, 13].
2-3. INDICATION ET MODE D'UTILISATION
L'IFO est largement utilisé en cancérologie [14] pour le traitement en première
intention, pour traiter les cas de rechute ou encore pour les cas de tumeurs
résistantes au traitement par d'autres agents. Il est classiquement administré, en
association à d'autres agents anticancéreux, dans les protocoles de traitement des
tumeurs du testicule [15], séminomateuses ou non séminomateuses, des cancers
pulmonaires à petites cellules [16, 17] ou à non-petites cellules [18], des lymphomes
hodgkiniens ou non hodgkiniens [19], des sarcomes des tissus mous [20, 21], de
cancer du sein [22], de cancer de l'utérus [23], de cancer de l'ovaire [24, 25] et de
cancer de l'os [26].
L'IFO en oncologie pédiatrique est indiqué dans les cas de résistance au CP, en
raison de sa moindre toxicité systémique (notamment la myélotoxicité) et de
l'absence de résistance croisée entre les deux médicaments. Dans les années 1980,
l'association IFO et mesna a été tentée chez les enfants surtout lors des rechutes ou
pour les tumeurs résistantes à la chimiothérapie. Il a été rapidement utilisé en
première intention dans de nombreuses tumeurs solides : sarcome d'Ewing,
lymphome, neuroblastome, médulloblastome, ostéosarcome, rhabdomyosarcome,
sarcome des tissus mous, et tumeur de Wilms [14, 27-29].
L'IFO peut être utilisé seul mais il est le plus souvent accompagné d'autre agents
anticancéreux, notamment, le cisplatine, l'étoposide, l'actinomycine D, la
doxorubicine ou la vincristine. Les protocoles sont nombreux, comportant des doses
de 1.8 à 5 g/m2 par jour sur 2 à 5 jours pour une dose totale de 6 à 15 g/cure [14, 30-
33]. Il est administré par une voie intraveineuse, pendant 24 heures ou pendant 1-3
heures (perfusion courte), accompagné de mesna à une dose de 80-120% de la
dose d'IFO. L'administration de mesna est prolongée de 12-24 heures au delà de
celle de l'IFO. Une hyperhydratation (3000 ml/m2) est recommandée. Les cures sont
répétées toutes les 3 à 4 semaines [14]. Sur le plan pharmacocinétique, et en
mesurant les principaux métabolites actifs et toxiques de l'IFO, Brain et al. ont trouvé
des résultats équivalents quel que soit le mode d'administration d'une dose de 9 g/m2
d'IFO en monothérapie sur 3 jours (soit en perfusion courte de 3 heures, soit en
perfusion continue) [30].
2-4. EFFETS SECONDAIRES
L'administration d'IFO est accompagnée de quelques effets toxiques aigus :
neutropénie, thrombocytopénie, nausée, vomissement, alopécie, arythmie etc. Ce
sont les effets qui accompagnent les protocoles de chimiothérapie en général. Elle
est aussi associée à d'autres effets toxiques plus sévères menaçant le pronostic
vital : cystite hémorragique, néphropathie, encéphalopathie et toxicité cardiaque [34].
2-4-1. Cystite Hémorragique
L'acroléine, un des métabolites hépatiques de l'IFO est à l'origine de la cystite
hémorragique. Le contact direct de l'acroléine avec la muqueuse urothéliale conduit
à un œdème, une ulcération, une néovascularisation, une hémorragie et une nécrose
[35]. La cystite hémorragique se manifeste par une hématurie macroscopique et des
brûlures mictionnelles plus ou moins importantes.
Depuis l'utilisation de mesna, un composé thiol synthétique qui détoxique l'acroléine
(en formant des composés thio-éther stables et non toxiques), cette complication a
bien été contrôlée [36-38]. Une hydratation adaptée du patient et l'administration
simultanée et systématique du mesna préviennent cette complication et permettent
d'utiliser l'IFO à des doses plus élevées. D'autres agents ont été proposés pour le
traitement de la cystite hémorragique : l'amifostine [39], l'interleukin-11 [40] et la
2-4-2. Néphrotoxicité
La néphrotoxicité de l'IFO a été décrite par plusieurs auteurs [14, 42-45]. Bien que
les premiers cas aient été rapportés chez l'adulte, la néphrotoxicité de l'IFO domine
chez l'enfant. D'importantes variations inter individuelles sont observées concernant
le type d'atteinte, sa sévérité, sa réversibilité et sa survenue par rapport au cours du
traitement. Les tubules rénaux proximaux représentent la cible de cette
néphrotoxicité. Tous les segments du néphron peuvent être touchés conduisant à un
dysfonctionnement soit glomérulaire, soit tubulaire proximal ou tubulaire distal, soit la
combinaison de toutes ces atteintes [44, 46-49].
2-4-2-1. Manifestations cliniques :
L'incidence de la néphrotoxicité après le traitement par l'IFO est variable selon les
études et les patients [46, 49-52], allant des signes infra cliniques chez 40% des
enfants traités à des atteintes rénales sévères (insuffisance rénale chronique,
tubulopathie, acidose tubulaire et syndrome de Fanconi chez 5-10% des enfants)
[53-55]. L'atteinte tubulaire proximale est la plus fréquente et la plus importante [56]
et peut se manifester par une diminution des taux de réabsorption des phosphates,
du bicarbonate, du glucose et des acides aminés [44, 45]. La lésion rénale pourrait
progresser même après l'arrêt du traitement [44, 46, 57]. Les séquelles cliniques les
plus connues sont l'hypophosphatémie et l'acidose tubulaire rénale qui peuvent
perturber la croissance ou entraîner un rachitisme hypophosphatémique [45, 55, 58,
59]. Skinner et al. ont montré que la néphrotoxicité était légère chez 50%, modérée
chez 20% et sévère chez 8% de leurs patients évalués après traitement par l'IFO
[54]. Le syndrome de Fanconi résulte d'une dysfonction généralisée des tubules
rénaux proximaux et se manifeste cliniquement par une diminution du débit de
filtration glomérulaire, glucosurie, phosphaturie, bicarbonaturie, aminoacidurie, et
protéinurie [43, 44].
2-4-2-2. Données pathologiques :
Elles sont non spécifiques et montrent essentiellement une atrophie des tubules
proximaux. La néphrite interstitielle est peu fréquente [60, 61].
2-4-2-3. Les facteurs de risque :
Plusieurs facteurs de risque de la néphrotoxicité ont été mis en évidence [44, 46, 54,
62]. La co-administration d'autres néphrotoxique comme le cisplatine [47, 53, 61, 63]
et la dose cumulative d'IFO (la dose totale administrée d'IFO) [46, 49, 52, 54]
peuvent augmenter la sévérité de la néphrotoxicité. Une néphrectomie antérieure [44,
53, 61], une présence de dysfonctionnement rénal préalable, d'infiltration tumorale
[45] ou d'irradiation des loges rénales peuvent aussi majorer la néphrotoxicité. De
plus, des variations quantitatives interindividuelles du métabolisme de l'IFO peuvent
exister et déterminer le risque individuel. L'âge jeune du patient, considéré comme
un facteur de risque [45, 46, 52, 64], est désormais mis en question [50, 54, 65]. Le
mode d'administration de l'IFO (la durée de cure, la durée de perfusion) ne semble
pas être important comme facteur de risque. La réversibilité de la néphrotoxicité de
l'IFO est incertaine. Selon Skinner et al., une partie importante de cette
néphrotoxicité était présente 10 ans après la fin du traitement [44], mais la sévérité et
le pronostic variaient entre les patients.
2-4-2-4. Prévention :
Contrairement à la cystite hémorragique, le mesna ne peut pas prévenir la
néphrotoxicité de l'IFO [51, 66-69]. L'amifostine, un médicament qui donne des thiols
actifs après sa déphosphorylation par la phosphatase alcaline, a été proposée pour
réduire la néphrotoxicité de l'IFO. Hartmann et al. ont trouvé que le débit de filtration
glomérulaire a été bien maintenu chez les patients recevant l'amifostine. Ils ont aussi
trouvé une baisse de 30% du débit de filtration glomérulaire chez les patients traités
sans recevoir l'amifostine. L'atteinte tubulaire était moins importante chez le groupe
d'amifostine [70]. Cependant, d'autres études ont montré l'absence de cet effet
protecteur d'amifostine contre la néphrotoxicité de l'IFO chez les enfants [71, 72], ou
chez le rat [69], malgré la présence de cet effet protecteur in vitro dans certaines
Dans un travail récent réalisé in vivo sur des rats, Chen et al. ont proposé que la N-
acétylcysteine (largement utilisée comme un antidote pour l'intoxication par le
paracétamol et l'acétaminophène chez les enfants) pourrait avoir un effet
thérapeutique éventuel contre la néphrotoxicité chez les enfants traités par l'IFO [74].
Ils avaient aussi montré que la N-acétylcysteine, appliquée in vitro sur des cellules
rénales, protège ces cellules contre la toxicité de l'IFO, probablement par
l'augmentation de la synthèse du glutathion [75].
2-4-2-5. Mécanismes de la néphrotoxicité de l'IFO :
Les mécanismes sous-jacents aux dommages rénaux induits par l'IFO ne sont pas
totalement élucidés. L'IFO en lui-même, n'est pas néphrotoxique [76-78]. Le CAA est
considéré comme le responsable de cette néphrotoxicité [45, 73, 76, 79, 80]. Les
mécanismes seront détaillés ultérieurement.
2-4-3. Encéphalopathie et neuropathie
L'IFO et ses métabolites pénètrent la barrière hémato-encéphalique et se trouvent
dans le liquide céphalorachidien en concentrations aussi importantes que dans le
sang [81]. La présence des ces métabolites produit des effets toxiques sur le
système nerveux central chez 10-30 % des patients traités. Cette encéphalopathie
se manifeste par des symptômes comme l'ataxie cérébelleuse, la confusion mentale,
les hallucinations visuelles, les signes extrapyramidaux, le mutisme et par des effets
moins fréquents comme l'astérixis, l'état épileptique non convulsif ou la dégénération
du cortex cérébral.
Les mécanismes physiopathologiques responsables de la neurotoxicité de l'IFO ne
sont pas complètement connus. Un déficit de l'oxydation mitochondriale des acides
gras résultant de l'inhibition du transfert d'électrons aux flavoprotéines avait été
proposé par Küpfer et al. [82]. Un autre mécanisme serait l'accumulation du CAA
dans le système nerveux central [83]. Un troisième mécanisme est basé sur la
neurotoxicité par l'intermédiaire de monoamine oxydase, dont l'inhibition par le bleu
de méthylène conduit à l'amélioration des signes cliniques et à la prévention de la
neurotoxicité [84]. D'autres études confirment l'efficacité du bleu de méthylène dans
le traitement et la prophylaxie de la neurotoxicité [83, 85, 86]. Hamadani et al. ont
proposé la thiamine pour traiter l'encéphalopathie induite par l'IFO [87].
2-4-4. Cardiotoxicité
Elle est de faible importance avec la dose standard d'IFO. Avec des fortes doses
d'IFO, la myocardite, la péricardite exsudative, la dépression myocardique et
l'insuffisance cardiaque congestive peuvent apparaître [88].
2-5. METABOLISME ET PHARMACOLOGIE
2-5-1. Généralités
L'IFO et le CP sont des isomères structuraux ne différant que par la position d'un
groupe chloroéthyl (Figure 6). Cette différence explique une meilleure solubilité de
l'IFO en milieu aqueux et une moindre toxicité systémique [20].
Figure 6 : La différence structurelle entre l'IFO et le CP.
Tous les deux sont des promédicaments qui doivent être activés par le système de
cytochromes P450 (CYP450) dans le foie. Leur activation aboutit à la formation des
moutardes azotées, les métabolites actifs, qui alkylent l'ADN des cellules
cancéreuses et provoquent leur mort par apoptose.
Bien que le métabolisme de l'IFO et du CP ait lieu dans le foie, d'autres organes
contenant les CYP450 peuvent participer à ce métabolisme. Il a été montré que les
cellules rénales sont capables de métaboliser l'IFO en 2- et 3-déchloroéthyl-IFO,
autrement dit, le CAA est produit localement par le tissu rénal [42, 89-91]. Le
métabolisme rénal de l'IFO pourrait contribuer à sa néphrotoxicité [42, 90].
2-5-2. Le Métabolisme hépatique
Le métabolisme hépatique de l'IFO passe par deux voies métaboliques : la 4-
hydroxylation (activation), et la N-déchloroéthylation (désactivation) (Figure 7).
1- L'IFO est activé par l'hydroxylation du quatrième carbone du cycle
d'oxazaphosphorine, une réaction catalysée par les CYP450 notamment CYP3A4,
CYP2B6 et CYP3A5 pour former l'intermédiaire instable 4-hydroxy ifosfamide (4-OH-
IFO) [13, 20, 92].
Figure 7: Schéma du métabolisme hépatique de l'IFO.
Le 4-OH-IFO se trouve en équilibre avec l'aldoifosfamide, son tautomère aldéhyde.
L'aldoifosfamide se décompose spontanément par
β -élimination en ifosforamide
moutarde, métabolite alkylant actif, avec la libération équimolaire de l'acroléine, un
aldéhyde hautement réactif, et responsable de l'urotoxicité. Le 4-OH-IFO peut être
métabolisé en 4-kétoifosfamide par l'ALDH. De même, l'aldoifosfamide peut être
transformé en carboxyifosfamide, une réaction également catalysée par l'ALDH. Le
4-kétoifosfamide et le carboxyifosfamide sont des métabolites inactifs.
2- La deuxième voie métabolique est la N-déchloroéthylation. Elle conduit à
l'inactivation de l'IFO par l'enlèvement d'un groupe chloroéthyl pour donner les
métabolites non toxiques 2- et 3-déchloroéthyl-IFO avec la production équimolaire du
CAA. La N-déchloroéthylation est également catalysée par les CYP P450,
notamment les CYP3A4 et CYP2B6 responsables de 71 % et 27 % du métabolisme,
respectivement [13].
2-5-3. Les Différences du métabolisme
Le métabolisme de l'IFO, et des oxazaphosphorines en général, est affecté par de
nombreux facteurs liés au produit lui-même (l'existence de formes racémiques), au
mode d'administration (voie d'administration, dose administrée, combinaison avec
d'autres médicaments et l'auto-induction) et aux patients (l'âge, le sexe, la fonction
rénale et hépatique et les facteurs génétiques).
Puisque l'IFO et le CP sont des isomères structuraux différant seulement par la
position d'un groupe chloroéthyl (Figure 6), il est remarquable que la néphrotoxicité
du CP soit rare. Suite à la différence structurelle entre l'IFO et le CP, des différences
de leur métabolisme, leur toxicité et leur spectre thérapeutique peuvent exister [20,
93]. Le CP est majoritairement métabolisé en 4-hydroxy-CP et seulement 5-10 % de
sa dose thérapeutique est métabolisé en CAA par la N-déchloroéthylation.
Contrairement, 20-40 % de la dose thérapeutique d'IFO est transformée en CAA [13,
94, 95]. Etant donné que le CAA est considéré comme le responsable de la
néphrotoxicité de l'IFO, cela pourrait expliquer l'incidence plus importante de cette
toxicité chez les patients traités par l'IFO comparés à ceux traités par le CP.
Il a été montré que les concentrations sanguines des métabolites actifs de l'IFO sont
trois fois moins élevées que celles obtenues à partir du CP après l'administration
intraveineuse de doses identiques d'IFO et de CP [96]. Cela peut être dû à l'affinité
inférieure de l'IFO pour certaines enzymes de bioactivation par rapport au CP. Il faut
donc appliquer l'IFO à une dose trois fois plus élevée que la dose de CP pour obtenir
les mêmes concentrations des produits actifs. Par conséquent, il y aura une
production des métabolites de la déchloroéthylation de l'IFO (en particulier le CAA)
beaucoup plus élevée qu'avec le CP [36, 97] ce qui amplifie encore la néphrotoxicité
de l'IFO par rapport à son analogue structurel.
La myélotoxicité de l'IFO est moins importante que celle du CP et cela permet
l'administration de doses plus élevées d'IFO [13, 14, 98]. L'IFO a une efficacité
thérapeutique supérieure à celle du CP [98-100]. Il pourrait être efficace même dans
les cas de résistance au CP [14]. La génération de CAA pourrait expliquer la
différence d'efficacité antitumorale entre l'IFO et le CP, ainsi que l'absence de
résistance croisée complète entre ces deux médicaments [101].
2-5-4. Le métabolisme énantiosélectif de l'IFO
L'IFO est un produit chiral. Sa molécule contient un atome asymétrique de
phosphore et donc il existe deux énantiomères : le R-IFO et le S-IFO. Il est
administré en tant que mélange racémique avec une distribution égale des deux
énantiomères. Le métabolisme du R-IFO et du S-IFO n'est pas identique. Il conduit à
la production des métabolites R ou S ayant une cytotoxicité différente.
Les cytochromes P450 dans le foie (notamment le CYP3A4, le CYP3A5 et le
CYP2B6) métabolisent les deux énantiomères. Il existe une préférence
énantiosélective dans le métabolisme, les pharmacocinétiques et l'efficacité
thérapeutique des deux énantiomères [102, 103]. Les CYP3A métabolisent le R-IFO
tandis que les CYP2B6 métabolisent le S-IFO [102, 103]. Donc, le R-4-hydroxy IFO,
le R-N2-déchloroéthyl IFO et le R-N3-déchloroéthyl IFO sont produits par les
CYP3A4 et 3A5, tandis que le S-4-hydroxy IFO, le S-N3-déchloroéthyl IFO et le S-
N2-déchloroéthyl IFO sont formés par le CYP2B6 [92, 103, 104].
Wang et al. [105] ont trouvé dans des études réalisées in vivo et in vitro que le
métabolisme du R-IFO passe d'une manière sélective par la 4-hydroxylation, tandis
que le S-IFO est métabolisé par la 2-N et 3-N déchloroéthylation. D'autres études
réalisées in vitro par Chen et al. [102] ont montré que les cellules tumorales
exprimant le CYP3A4 métabolisent le R-IFO pour obtenir le 4-hydroxy IFO, de façon
plus importante que le S-IFO contrairement aux cellules exprimant le CYP2B. Ils ont
proposé que le R-IFO puisse avoir une activité anticancéreuse plus importante que le
S-IFO et le CP contre les tumeurs possèdant les CYP3A4.
2-5-5. Le métabolisme rénal de l'IFO
Pendant ces dernières années, plusieurs auteurs ont travaillé sur le métabolisme
tubulaire rénal de l'IFO et sur la production rénale locale du CAA.
Dans un modèle de rein isolé et perfusé chez le rat, Springate et al. ont montré la
production rénale de 2-N et 3-N déchloroéthyl IFO [90]. Il a été également montré
que les cellules rénales humaines et porcines possèdent dans leur microsomes les
deux cytochromes CYP3A4 et CYP2B6 nécessaires pour le métabolisme de l'IFO et
la production du CAA, le néphrotoxique présumé de l'IFO [42, 64, 78, 91]. Aleksa et
al. ont démontré que les concentrations de CAA produites dans les tubules rénaux
étaient comprises entre 35 et 320 µM (moyenne de 80 µM) ce qui correspond aux
concentrations néphrotoxiques [106]. Cela suppose que la néphrotoxicité de l'IFO
pourrait être due, au moins en partie, à la production locale du CAA dans le cortex
rénal. Les CYP3A4 et CYP3A5 sont présents dès la huitième semaine de grossesse
et sont capables de métaboliser les deux énantiomères de l'IFO. La quantité totale
de CYP rénal est environ six fois moindre que celle de CYP hépatique [89]. La
contribution rénale dans le métabolisme de l'IFO reste donc inférieure à celle du foie.
En outre, Aleksa et al. ont montré que les cellules tubulaires rénales métabolisent les
deux énantiomères R et S de l'IFO en leurs métabolites 2- et 3-déchloroéthyl IFO
[42]. Ils ont aussi montré un métabolisme rénal énantiosélectif de l'IFO avec une
production plus importante de N-déchloroéthyl IFO à partir de S-IFO par rapport à la
production de N-déchloroéthyl IFO à partir de R-IFO. Ils proposent que le
remplacement du mélange racémique d'IFO par le R-IFO seul pourrait diminuer le
métabolisme rénal de l'IFO en N-déchloroéthyl IFO (donc en CAA) et par conséquent,
diminuer sa néphrotoxicité [107].
2-6. MECANISMES DE LA NEPHROTOXICITE DE L'IFO. DONNEES
La responsabilité du CAA dans la toxicité rénale lors du traitement par l'IFO est
suggérée du fait des particularités pharmacologiques de l'IFO et du CP. Etant donné
que la néphrotoxicité accompagne uniquement le traitement par l'IFO et non par le
CP, et que le métabolisme de l'IFO produit des quantités de CAA plus élevées que le
métabolisme du CP, le CAA est incriminé dans la néphrotoxicité de l'IFO. Par ailleurs,
les variations interindividuelles importantes du métabolisme de l'IFO pourraient
conduire à une production plus ou moins importante du CAA pouvant expliquer les
susceptibilités individuelles à la toxicité rénale d'IFO observée en pratique clinique.
Récemment, Nissim et al. [43] ont montré, après un traitement quotidien de 50 mg/kg
d'IFO pendant 5 jours chez les rats, que la concentration de CAA dans le cortex
rénal était de 15 µM, ce qui confirme l'accumulation de ce toxique dans le cortex
rénal, pouvant être à l'origine de la néphrotoxicité. D'ailleurs, la présence des
enzymes CYP appropriées dans les cellules tubulaires rénales et leur capacité à
métaboliser l'IFO en ses métabolites 2- et 3-déchloréthyl IFO porte à croire que la
lésion néphrotoxique pourrait être également liée à la production locale de CAA.
La détection de CAA est difficile à cause de son instabilité. Il est rapidement
métabolisé en chloroacétate par l'ALDH. Par conséquent, sa concentration est
souvent déterminée par les mesures des deux produits stables du métabolisme de
l'IFO (le 2- et 3-déchloroéthyl IFO) car le CAA est produit en quantité équimolaire à
celle des deux produits [106]. La demi-vie du CAA chez l'homme in vivo est courte :
2-8 heures [108]. Les concentrations sanguines et urinaires de CAA mesurées chez
les patients traités par l'IFO sont variables entre 5 et 200 µM selon les études [33, 96,
1- L'effet toxique de 50 µM CAA a été montré sur des cellules tubulaires rénales en
culture par Mohrmann et al. [112] (diminution de contenu des protéines cellulaires) et
par Zaki et al. [73] avec 25-200 µM CAA (baisse du glutathion et du transport du
2- Il a été aussi montré sur un modèle de rein de rat isolé et perfusé que 210 µM
CAA perturbe la fonction rénale : inhibition de la réabsorption tubulaire de sodium,
glucose, phosphate et sulfate inorganiques, ce qui indique un dysfonctionnement
tubulaire proximal généralisé [78].
3- Dubourg et al. ont montré que 500 µM CAA avaient un effet toxique sur les tubules
rénaux proximaux humains se manifestant par une diminution d'ATP et du glutathion
et une augmentation de la libération de LDH. Ils ont aussi montré que ces tubules
sont capables de métaboliser, et ainsi détoxiquer le CAA, probablement par l'ALDH,
une enzyme très active au niveau du rein humain. La RMN de 13C avait montré que
ces tubules transforment le CAA en chloroacétate, un métabolite du CAA beaucoup
moins toxique [113].
4- Springate et al. ont démontré, dans une étude réalisée sur des rats in vivo, que le
CAA induit un dysfonctionnement rénal (manifesté par une diminution du débit de
filtration glomérulaire, une augmentation du débit urinaire et une augmentation de la
sécrétion du glucose, du sodium et des protéines), une déplétion du glutathion et une
augmentation de la péroxydation des lipides. Les concentrations sanguines initiales
de CAA étaient comprises entre 100 et 900 µM [80].
5- La même équipe a trouvé, en appliquant le CAA à 12.5-150 µM sur des cellules
rénales proximales en culture, une diminution dose dépendante d'ATP, du glutathion
et de la croissance cellulaire. L'IFO lui-même n'était toxique qu'à très forte
concentration (4000-10000 µM) [77]. Ils ont récemment remontré cet effet toxique en
appliquant le CAA à 6.25-100 µM sur le même type de cellules [114].
2-7. CIBLES DE LA NEPHROTOXICITE DU CAA
Les mécanismes exacts de la néphrotoxicité induite par le CAA ne sont pas
complètement connus.
1- Le CAA induit une chute d'ATP cellulaire [77, 113]. Il conduit également à une
diminution du glutathion [73, 77, 80, 113], des thiols cellulaires [77, 79, 115], du
coenzyme A et d'acétyl coenzyme A [115]. Le CAA augmente également la
péroxydation des lipides [77].
Le glutathion joue des rôles importants dans de nombreuses fonctions biologiques, y
compris la détoxication, la protection du stress oxydatif et la maintenance du
potentiel redox cellulaire [73]. En effet, le CAA réagit avec les groupes sulfhydryl
(SH) des protéines membranaires et intracellulaires pour former, via une réaction
chimique non enzymatique, des adduits conduisant à endommager les cellules [116].
La substitution en glutathion empêcherait les dégâts cellulaires induits par le CAA.
Les molécules contenant des groupes SH protègent les tubules proximaux en
réagissant avec le CAA avant que ce dernier puisse perturber d'autres groupes SH
importants dans la cellule. Par conséquent, le glutathion peut protéger les cellules,
non seulement par son rôle d'antioxydant, mais également en fournissant des
groupes SH qui se lient au CAA [76].
2- Le CAA provoque la mort cellulaire plutôt par nécrose que par apoptose. Benesic
et al. [79] ont trouvé, dans une étude réalisée sur des cultures de tubules rénaux
proximaux humains, que la réduction du nombre des cellules et des protéines
cellulaires est accompagnée d'une augmentation des marqueurs de la nécrose (la
libération de LDH et la capture de bleu de Trypan) et cela d'une façon dose
dépendante. Ces observations sont également décrites dans d'autres publications
[76, 117, 118]. Le CAA, en modifiant les groupes catalytiques SH, inhibe l'activité des
caspases. Les caspases sont des enzymes indispensables pour l'apoptose cellulaire.
Une cascade des réactions conduit à l'activation de caspase-3 qui orchestre le
processus d'apoptose. L'inhibition de cette cascade pourrait être un mécanisme de la
nécrose induite par le CAA [79].
3- Un autre mécanisme d'action du CAA est proposé par Benesic et al. Ils ont montré
que le CAA (1.5-150 µM) induit une augmentation des concentrations intracellulaires
du calcium libre ([Ca2+]i) dans des tubules rénaux proximaux [117]. Cela résulte de
l'inhibition de l'échangeur sodium/calcium. Une liaison covalente entre le CAA et la
protéine de l'échangeur lui-même ou une autre protéine kinase impliquée dans son
fonctionnement, semblerait être à l'origine de cette inhibition [117].
4- L'inhibition du transport sodium dépendant du phosphate est un autre effet du
CAA (50-100 µM/24h) sur les cellules rénales, la réduction de l'expression de la
protéine NaPi-IIa étant à l'origine de cet effet [118]. Mohrmann et al. avaient déjà
montré que l'exposition des cellules rénales en culture au CAA détériore plusieurs
systèmes de transport cellulaire sodium dépendant ainsi que le système antiport
sodium/proton [119, 120].
5- Récemment, l'effet du CAA sur l'oxydation mitochondriale a été étudié par Nissim
et al. [43]. Le CAA, à une concentration de 1000 µM, a inhibé la phosphorylation
oxydative au niveau du complexe I de la chaîne respiratoire dans des mitochondries
fraîchement isolées du cortex rénal. Dans leurs expériences, le toxique avait été
appliqué directement aux mitochondries isolées. L'inhibition de la respiration
mitochondriale avait également été montrée dans des hépatocytes isolées [121].
3. PHYSIOLOGIE DU CANCER
3-1. HISTORIQUE DU METABOLISME DU CANCER
Le métabolisme du glucose dans les cellules cancéreuses est altéré. Dans la plupart
des cellules des mammifères, la glycolyse est inhibée par la présence d'oxygène, qui
permet aux mitochondries d'oxyder le pyruvate en CO2 et H2O. Cette inhibition est
appelée «l'effet Pasteur» car Louis Pasteur a été le premier à le démontrer [122].
L'inhibition de la glycolyse est essentielle pour assurer la production d'énergie
lorsque l'oxygène est présent. La conversion du glucose en lactate même en
présence d'oxygène est connue sous le nom de glycolyse aérobie et est appelée
«l'effet Warburg». C'est Otto Warburg qui a réalisé les premiers travaux sur le
métabolisme du cancer en 1929 et qui a résumé ses résultats en 1956 [123].
L'augmentation de la glycolyse aérobie est observée dans les cellules cancéreuses
et les cellules transformées et non dans les cellules normales. Warburg a donc
proposé l'hypothèse que le cancer est le résultat d'un dysfonctionnement de la
respiration mitochondriale, compensée par une contribution élevée et exceptionnelle
de la glycolyse aérobie dans le turnover de l'ATP total. Bien que cette hypothèse se
soit avérée incorrecte, la perception du changement métabolique de la
phosphorylation oxydative à la glycolyse aérobie dans les cellules tumorales, reste
toujours d'actualité [124-127].
3-2. MECANISME DE L'EFFET WARBURG
De nombreux mécanismes pourraient être responsables de l'augmentation de la
glycolyse dans les cellules cancéreuses [128, 129].
3-2-1. Mutations ou suppression de l'ADN mitochondrial
Il est possible que les mutations, largement observées dans les cellules cancéreuses,
puissent produire un dysfonctionnement mitochondrial et une diminution de la
capacité cellulaire à générer l'ATP via la phosphorylation oxydative [130, 131].
Plusieurs facteurs contribuent à l'augmentation de taux des mutations au niveau de
l'ADN mitochondrial. Il est très proche des sites de génération des molécules
oxygénées réactives (ROS pour reactive oxygen species) dans les mitochondries, il
a une faible capacité à se réparer et il n'est pas protégé par les histones [129]. Le
génome mitochondrial encode 13 composants protéiques importants de la chaîne
respiratoire. Les mutations d'ADN pourraient endommager ses protéines, et par la
suite, perturber la fonction de la chaîne respiratoire. Le dysfonctionnement de la
chaîne respiratoire oblige les cellules à utiliser la glycolyse pour produire l'ATP. Etant
donné que la production d'ATP par les mitochondries est beaucoup plus efficace que
celle par la glycolyse (36 ATP par la phosphorylation oxydative contre 2 ATP par la
glycolyse, à partir d'un glucose), une petite perte de la fonction respiratoire
entraînerait une augmentation importante de la glycolyse afin de maintenir la balance
Lors de l'exposition à l'oxygène, la plupart des cellules cancéreuses montrent une
consommation d'oxygène plus ou moins importante, ce qui suppose que la fonction
mitochondriale est conservée. Dans les cellules ayant un taux de croissance rapide,
la génération d'ATP par la phosphorylation oxydative est active et elle fournit une
partie importante de l'énergie totale [132]. Nous pouvons donc dire que la glycolyse
n'est pas la source unique de la génération d'ATP par les cellules cancéreuses.
3-2-2. Rôle du microenvironnement : l'hypoxie
L'hypoxie est un modulateur puissant du métabolisme énergétique. Elle impose
l'utilisation de la glycolyse par les cellules cancéreuses pour générer l'ATP, même en
l'absence de mutations mitochondriales. C'est le cas pour les cellules de cancer qui
se trouvent dans un environnement pauvre en oxygène. L'hypoxie est fréquente
dans les cancers humains, surtout les tumeurs solides, quand la croissance tumorale
atteint un volume important qui limite la pénétration d'oxygène. Dans ces conditions,
la phosphorylation oxydative ne fonctionne pas normalement à cause de
l'insuffisance d'oxygène même en absence des défauts structurels des
L'augmentation de la glycolyse conduit à une production élevée du lactate qui
conduira à l'acidification des tissus tumoraux, créant le microenvironnement
caractéristique du cancer.
L'augmentation de la glycolyse peut être vue comme une adaptation à l'hypoxie [125].
La persistance de la glycolyse même en présence d'oxygène, est une façon
d'adaptation à l'hypoxie intermittente dans les lésions précancéreuses. La
surexpression de la glycolyse conduit à un microenvironnement acide qui exige
l'évolution des phénotypes résistant à l'acidité. Les générations suivantes des
cellules qui ont une surexpression de la glycolyse et une résistance à l'acidité,
possèdent une puissante capacité de croissance, de prolifération et d'invasion [125].
Rôle du facteur inductible par l'hypoxie (HIF-1)
Le facteur inductible par l'hypoxie (HIF-1 pour hypoxia inducible factor) a été identifié
au début des années 1990 comme un facteur de transcription. C'est un complexe
composé de deux sous unités : HIF-1β qui est surexprimé en permanence, et HIF-1α
dont le niveau dépend de la concentration d'oxygène dans le tissu. HIF-1β a été
identifie comme étant le facteur ARNT1 (aryl hydrocarbon receptor nuclear
translocator), une protéine déjà décrite et impliquée dans les phénomènes de
détoxification des cellules. En revanche, HIF-1α est spécifique de la réponse
hypoxique. Dans les cellules bien oxygénées, HIF-1α a une demi-vie courte et se
dégrade rapidement par le système ubiquitine-protéase. Il est donc non détectable
dans les conditions de normoxie mais il augmente rapidement lors de l'hypoxie [133].
L'HIF est souvent surexprimé dans les cellules de cancer. La surexpression est
accompagnée d'un mauvais pronostic et d'une mauvaise réponse à la chimio ou à la
radiothérapie. La régulation positive de ce facteur peut être également responsable
de la résistance et de la baisse de sensibilité des tumeurs à la radiothérapie,
fréquemment associées à l'hypoxie [129].
L'HIF-1, ainsi que d'autres éléments de la glycolyse, peuvent être activés
directement par des mutations dans les cellules cancéreuses indépendamment de
l'hypoxie. En effet, l'activation d'HIF-1 peut être la conséquence d'une hypoxie locale
ou d'autres facteurs indépendants de l'oxygène comme : l'inactivation de nombreux
gènes suppresseurs de tumeurs, l'activation de plusieurs oncogènes et l'activation
des facteurs de croissance [126, 134].
La réponse cellulaire à l'hypoxie est contrôlée par l'HIF-1 [135] qui active l'expression
des gènes impliqués dans le transport et le métabolisme du glucose et les gènes
impliqués en cancérogenèse : l'angiogenèse, la captation du glucose, les signaux
des facteurs de croissance, l'apoptose, l'invasion et les métastases [134, 136].
L'importance du rôle d'HIF-1 est citée par Greijer et al. qui considèrent que le
changement métabolique vers la glycolyse, observé dans les cellules tumorales,
n'est pas une simple conséquence de l'hypoxie et du blocage de la phosphorylation
oxydative, mais un programme transcriptionnel initié par l'HIF-1[137].
L'HIF-1 stimule l'expression d'environ toutes les enzymes de la glycolyse ainsi que le
transporteur de glucose GLUT-1 [138]. En plus, il inhibe le métabolisme du pyruvate
et la consommation d'oxygène par les mitochondries. Il inhibe ainsi le cycle des
acides tricarboxyliques et la respiration en induisant la pyruvate déshydrogénase
kinase qui inhibe l'activité pyruvate déshydrogénase (PDH) par phosphorylation. Le
pyruvate n'entre plus dans le cycle des acides tricarboxyliques et s'accumule avant
d'être transformé en lactate [139, 140].
3-2-3. La signalisation oncogène
La signalisation oncogène, qui contrôle la transformation maligne des cellules, peut
également réguler le métabolisme énergétique et rendre les cellules cancéreuses
hautement glycolytiques et plus dépendantes de la glycolyse que de l'oxydation
mitochondriale pour produire l'ATP. La protéine Ras, l'Akt kinase et le c-myc sont
parmi les molécules impliquées dans la signalisation oncogènes [141].
3-2-4. Mutations ou altérations d'expression génétique d'ADN nucléaire
C'est un autre mécanisme du dysfonctionnement mitochondrial car l'ADN nucléaire
est impliqué dans le codage de la majorité des composants protéiques
3-2-5. Altérations d'expression enzymatique
Elles induisent des changements métaboliques dans les cellules cancéreuses
conduisant à l'effet Warburg. Par exemple, l'expression du gène de la GAPDH est
associée à la prolifération des cellules de cancer du sein et associée également à
l'agressivité des tumeurs [142].
3-3. ROLE DE L'EFFET WARBURG DANS LA CARCINOGENESE
L'effet Warburg apporte des avantages aux cellules cancéreuses dans lesquelles cet
effet se manifeste [143].
1- La biosynthèse : les cellules cancéreuses ayant une grande vitesse de croissance
ont besoin de précurseurs carbonés pour la biosynthèse. La voie glycolytique et la
voie des pentoses phosphate représentent une source riche de précurseurs
nécessaires pour la synthèse des macromolécules : les acides nucléiques, les
phospholipides, les acides gras, le cholestérol, les porphyrines et d'autres
composants nécessaires pendant la division cellulaire [144]. Une des principales
fonctions de la glycolyse dans les cellules proliférantes est donc de fournir les
précurseurs pour la synthèse de ces macromolécules [141, 145-147].
2- Protection et invasion : les cellules cancéreuses produisent, par la glycolyse, du
lactate qui est transporté en dehors des cellules. Ceci crée un milieu extracellulaire
acide qui protège les cellules tumorales, résistantes à l'acidité, contre les attaques du
système immunitaire et induit des effets chimiques négatifs sur les cellules normales
autour de la tumeur pour préparer l'invasion tumorale [125].
3- L'effet Warburg assure également la survie des tumeurs quand l'oxygène devient
limitant. Les enzymes qui catalysent la voie glycolytique, déjà induites, ne sont pas
dépendantes de l'oxygène. Au contraire, les gènes qui encodent ces enzymes sont
activés par l'hypoxie via l'HIF-1.
3-4. MODIFICATION DU METABOLISME DU GLUCOSE DANS LES
L'augmentation de la glycolyse dans les cellules tumorales, décrite par Warburg,
pourrait être expliquée par des modifications enzymatiques et par une augmentation
du transport transmembranaire et de la captation cellulaire du glucose. La part
relative de ces deux phénomènes, qui peuvent être perturbés de façon dissociée,
varie d'une tumeur à l'autre. Ces phénomènes ne traduisent pas un dérèglement
anarchique du métabolisme tumoral, en réaction aux conditions environnementales
difficiles des tumeurs, mais résultent d'une programmation génétique parfaitement
ordonnée. Ce programme reproduit de façon permanente les mécanismes
physiologiques d'adaptation métabolique mis en jeu lors de la prolifération des tissus
sains: la croissance, la cicatrisation ou le renouvellement tissulaire. Ces
modifications donneraient un avantage sélectif aux cellules tumorales. La mauvaise
qualité du microenvironnement, sans être le facteur causal, intervient comme
pression de sélection des cellules à capacité glycolytique élevée [148].
3-4-1. Modifications des enzymes clés de la glycolyse
Dans les cellules tumorales, une perte de l'effet Pasteur (inhibition de la glycolyse
par la production mitochondriale d'ATP) et un niveau élevé de glycolyse en
conditions aérobies sont observées [123, 149]. L'augmentation de la glycolyse
résulte des modifications enzymatiques quantitatives et qualitatives qui se
manifestent par une augmentation des activités enzymatiques dans les tissus
tumoraux. Cette augmentation est encore plus remarquée dans les tumeurs
agressives ou ayant des capacités métastatiques élevées [149, 150]. Les
phénomènes les plus importants concernent les trois enzymes clés : HK, PFK et PK
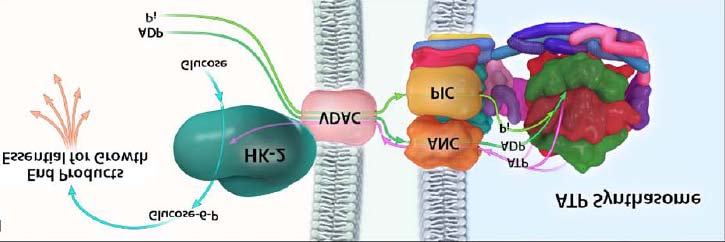
Environ 75% de l'HK des cellules tumorales est liée à la membrane externe des
mitochondries [151] via un récepteur VDAC (Voltage Dependent Anion Chanel),
(Figure 8). Elle est couplée directement à l'ATP synthase liée à la membrane interne,
ce qui fournit l'ATP nécessaire pour le fonctionnement de l'enzyme et favorise le
catabolisme du glucose en déclenchant la première étape de la glycolyse [143, 152].
Il s'agit de l'hexokinase II (HK-II), une des quatre isoenzymes de la famille
hexokinase. Elle a une haute affinité pour le glucose. Son association réversible aux
mitochondries est un mécanisme important de régulation de son activité : la forme
liée n'est pas (ou très peu) sensible à l'inhibition par le G6-P comparativement à la
forme soluble [153]. L'HK-II est abondante dans plusieurs types de cellules
tumorales [154, 155] et également dans le tissu de cancer du sein où son expression
n'est pas liée à celle du transporteur du glucose [156]. Rampel et al. avaient
également trouvé que l'HK-II était amplifiée 5 fois dans des cellules cancéreuses
ayant une haute activité glycolytique [157].
Figure 8: La liaison entre l'hexokinase et la membrane mitochondriale. D'après Pedersen. J
Bioenerg Biomembr (2007) 39 : 211-222.
Il existe une augmentation de l'activité PFK dans les tumeurs hépatiques [155]
variable en fonction du degré de différenciation et accompagnée d'une modification
du profil d'expression des isoenzymes [158]. Dans les cellules normales, l'effet
Pasteur est expliqué par l'inhibition de la PFK par l'ATP, le citrate et d'autres
métabolites. L'expression préférentielle dans les tumeurs de l'isoenzyme, qui n'est
pas sensible à ces métabolites [155], explique au moins en partie, la perte de l'effet
L'activité PK est très élevée dans les cellules tumorales [149, 159]. Dans des
tumeurs du sein, cette augmentation de l'activité PK est corrélée au degré de
malignité par référence au tissu mammaire sain. Cela est lié à l'expression d'une
forme modifiée, caractéristique des cellules embryonnaires et tumorales, la PK-M2
qui a été détectée dans les tumeurs du sein [159] et retrouvée dans les cellules
proliférantes et tumorales [160-162]. La PK-M2 existe sous deux formes : une forme
dimérique inactive, et une autre tétramérique très active ayant une affinité élevée
pour son substrat, le PEP, et liée au complexe des enzymes glycolytiques. La PK-M2
régule la proportion de glucose orientée vers les voies de biosynthèses (la forme
dimérique) ou utilisée pour la production d'énergie (la forme tétramérique). Le ratio
tétramère : dimère n'est pas stationnaire. Il oscille entre la forme tétramérique et la
forme dimérique sous la régulation d'ATP et du F1,6-P2. Quand le F1,6-P2
s'accumule, par exemple, la forme dimérique s'associe pour donner la forme active
tétramérique qui accélère la conversion du pyruvate en lactate et la production
d'énergie jusqu'au retour à la normale du F1,6-P2 [150, 162]. Les expériences
réalisées par Christofk et al. [160, 163] ont montré l'importance de la PK-M2 pour le
métabolisme et la croissance des tumeurs. Le basculement de l'expression
enzymatique de la PK-M2 à la PK-M1 (l'isoenzyme exprimée dans la plupart des
cellules adultes et normales) conduit à l'annulation de l'effet Warburg dans des
cellules cancéreuses. Cette annulation se manifeste par une diminution de la
production du lactate, une augmentation de la consommation d'O2, et une réduction
de la capacité de croissance de ces cellules [160]
3-4-2. Modifications de la voie des pentoses phosphate
La phosphorylation de la PK-M2 par les protéine kinases est inhibée par le F1,6-P2, le
phospho-ribose-pyrophosphate et le P1,P5-di(adénosine-5'-)-pentaphosphate ; des
composés dont la concentration s'élève après la stimulation hormonale de la
croissance cellulaire. Inversement, toute baisse de ces composés (qui se produit
lorsque le processus de prolifération perdure, ce qui est le cas des tumeurs)
déclenche la phosphorylation de la PK-M2 et son inhibition. Ce blocage relatif de la
PK augmente la concentration de GAP et favorise l'accumulation de ribose-5-
phosphate par la voie non oxydative des pentoses phosphate. Les taux élevés de
F1,6-P2, également rencontrés dans ces conditions, inhibent la 6-phosphogluconate
déshydrogénase, ce qui favorise la direction non oxydative de la voie des pentoses
phosphate. Ainsi, le frein relatif apporté par l'inactivation de la PK permet le maintien
du «compartiment de métabolites phosphorés» [149].
3-4-3. Augmentation du transport du glucose
Il est admis que la captation élevée du glucose par les cellules transformées et
tumorales est liée à une augmentation de son transport transmembranaire [164],
indépendamment de la phosphorylation et des étapes ultérieures de la glycolyse.
Nous observons en effet la même augmentation de captation en utilisant le 3-O-
méthyl-D-glucose (3-OMG) qui diffuse par le même transporteur mais qui n'est pas
phosphorylé par l'HK, ce qui confirme l'augmentation du transport transmembranaire
lui-même [165].
Le transport du glucose dans les cellules tumorales, qui représente la première étape
avant d'initier son métabolisme, est assuré par un transporteur présentant une stricte
analogie avec le transporteur GLUT-1 rencontré au niveau des hématies et dans la
plupart des tissus [166]. Le GLUT-1 est un membre de la famille GLUT, responsable
du transport facilité du glucose qui fonctionne selon le gradient de concentration
indépendamment de l'énergie. Treize membres de cette famille ont été identifiés
selon les différents tissus [167].
L'augmentation du nombre total de transporteurs est le mécanisme probable de
l'augmentation de transport du glucose en rapport avec le processus de
transformation et les cellules cancéreuses. Plusieurs études ont montré une
surexpression du GLUT-1 dans les cellules et tissus tumoraux par rapport aux tissus
normaux [156, 166-169]. Cette surexpression est associée à l'agressivité des
tumeurs et à leur capacité métastatique [170]. D'autres transporteurs peuvent être
impliqués dans le transport du glucose. Rogers et al. ont découvert un nouveau
transporteur (GLUT-12) dans les cellules MCF-7 de cancer du sein humain [171] et
dans des tumeurs du sein [172]. L'augmentation de GLUT-1 est génétiquement
déterminée. La transformation maligne est accompagnée d'une surexpression du
gène encodant le GLUT-1, qui se traduit par une augmentation des ARNm
correspondants [173]. L'augmentation de l'ARNm de GLUT-1 a été également
démontrée sur des cellules de cancers humains.
Plusieurs facteurs sont impliqués dans la régulation de l'expression de GLUT-1 dans
le cancer. L'hypoxie, via l'HIF-1, peut augmenter les niveaux de GLUT-1, et ainsi la
captation du glucose, dans des cellules ou des tissus cancéreux [174, 175].
L'estradiol est impliqué dans la régulation de l'expression de GLUT-1 et GLUT-12
[176, 177]. La captation cellulaire du glucose est stimulée par des facteurs de
croissance, notamment l'EGF (epidermal growth factor) [178, 179].
3-5. IMPLICATIONS THERAPEUTIQUES DE L'EFFET WARBURG.
DONNEES EXPERIMENTALES
L'effet Warburg fournit une base biochimique importante pour désigner des
stratégies et utiliser de nouveaux agents thérapeutiques dans le traitement du cancer
[128, 129]. Les cellules cancéreuses comptent sur la glycolyse pour générer l'ATP.
Ceci est une différence métabolique majeure entre les cellules malignes ou
proliférantes et les cellules normales qui peuvent utiliser d'autres sources d'énergie
comme les acides gras ou les acides aminés via le cycle des acides tricarboxyliques.
Les cellules normales seraient donc moins sensibles à l'inhibition de la glycolyse.
Cela a conduit au développement d'un concept thérapeutique basé sur l'inhibition de
la glycolyse pour tuer les cellules cancéreuses d'une façon préférentielle [129, 180-
185], ou pour améliorer l'efficacité et réduire la toxicité de certains agents de la
chimiothérapie [186, 187] (Figure 9).
La conversion du glucose en G6-P, catalysée par l'HK, est la première étape
limitante de la glycolyse. Etant donné que le G6-P est un métabolite intermédiaire
commun entre la voie de la glycolyse et la voie des pentoses phosphate (précurseur
des biosynthèses), l'inhibition de l'HK aurait des effets profonds sur les deux voies
métaboliques. Le 2-désoxyglucose, le 5-thioglucose (deux analogues du glucose), le
3-bromopyruvate et la lonidamine sont des inhibiteurs de l'HK.
1- Le 2-Désoxyglucose (2-DOG) est un analogue du glucose qui fonctionne comme
un inhibiteur compétitif du métabolisme du glucose. Une fois transporté dans les
cellules, il est phosphorylé par l'HK en 2-DOG-6-P qui n'est plus métabolisé et donc
s'accumule dans les cellules et inhibe l'HK, probablement par le rétrocontrôle négatif
de ce produit. A des concentrations millimolaires, le 2-DOG engendre une déplétion
d'ATP et la mort cellulaire in vitro, surtout dans les cellules cancéreuses qui ont des
mitochondries altérées ou qui se trouvent dans un environnement hypoxique [180,
188]. In vivo, le 2-DOG augmente l'efficacité des agents anticancéreux (l'adriamycine
et le placlitaxel) dans les souris porteuses des xénogreffes de cancer du poumon ou
d'ostéosarcome [187]. La forte concentration de 2-DOG nécessaire pour produire
son effet cytotoxique, et la présence de la compétition par le glucose déjà présent à
forte concentration, peuvent affecter l'efficacité thérapeutique prévue de ce composé.
Figure 9 : Inhibition de la glycolyse pour le traitement de cancer. D'après Chen et al. J
Bioenerg Biomembr (2007) 39 : 267-274.
2- Le 3-bromopyruvate est un pyruvate halogéné, un agent alkylant puissant contre
les groupes SH des résidus cystéine dans les protéines. Il peut affecter plusieurs
enzymes grâce à sa capacité alkylante. Des études ont démontré que le 3-
bromopyruvate est un inhibiteur de l'HK et de la respiration mitochondriale,
conduisant à la déplétion d'ATP et la mort cellulaire [189-191]. Le 3-bromopyruvate a
montré un effet anti-glycolytique important sur les cellules de cancer du sein
implantées chez les rats [192]. Il a montré également une activité anti-cancéreuse
prometteuse in vivo contre les cellules du carcinome hépatique sans toxicité
hépatique importante [189, 190]. Dans leur étude, Xu et al. ont montré que le 3-
bromopyruvate induit une déplétion sévère de l'ATP et la mort cellulaire quand il a
été appliqué sur des cellules cancéreuses ayant des défauts des mitochondries ou
sous conditions d'hypoxie (qui réduit habituellement la sensibilité des cellules aux
agents anti-cancéreux) [191]. Ils ont également montré que la déplétion d'ATP
induite par le 3-bromopyruvate peut désactiver les pompes (dépendantes d'ATP) qui
exportent plusieurs médicaments, tuant ainsi les cellules cancéreuses.
Le 3-bromopyruvate possède donc des propriétés anti-cancéreuses prometteuses et
peut être efficace contre des cancers hypoxiques ou résistants au traitement. Bien
que l'efficacité du 3-bromopyruvate soit supérieure à celle du 2-DOG, il est à noter
qu'il est instable dans les solutions et nécessite une concentration de 100 µM pour
3- La lonidamine est un dérivé de l'indazole-3-carboxylique capable d'inhiber la
glycolyse aérobie dans des cellules cancéreuses, mais pas dans les cellules
normales, probablement par l'inhibition de l'HK-II, liée aux mitochondries [193]. Elle
est efficace contre les cellules résistantes et sensibles à la chimiothérapie. Dans les
cellules MCF-7, la lonidamine peut également augmenter l'efficacité de quelques
agents alkylant comme le cisplatine, le 4-hydropéroxycyclophosphamide et le
melphalan [194].
La PFK catalyse la phosphorylation du F6-P en F1,6-P2, une étape limitante de la
glycolyse qui consomme de l'énergie. La PFK est régulée par l'ATP (régulation
négative) et par le F2,6-P2 (régulation positive). La suppression d'une isoenzyme de
la PFK (la B3) est proposée par Chesney et al. comme étant une stratégie pour tuer
les cellules transformées de rat d'une manière préférentielle [195]. Spitz et al. ont
montré que l'acide acétylsalicylique et l'acide salicylique inhibent la PFK dans les
cellules MCF-7 de cancer du sein, conduisant ainsi à une inhibition de la glycolyse et
la viabilité de ces cellules [185].
En plus de son rôle comme une enzyme impliquée dans le métabolisme du glucose,
la GAPDH affecte plusieurs fonctions cellulaires et nucléaires [196]. A part son rôle
dans l'apoptose, elle est importante pour le contrôle d'expression génétique des
histones, la formation des vésicules membranaires, la reconnaissance des
nucléotides mal incorporés dans l'ADN, le transport de l'ARN nucléaire, la réplication
et la réparation de l'ADN. Enfin, la GAPDH peut être directement impliquée dans le
phénotype des maladies neurodégénératives [197, 198]. Parmi les inhibiteurs de la
GAPDH, on peut citer : l'α-chlorohydrine, l'ornidazole, l'iodoacétate et des composés
3-5-4. Lactate déshydrogénase (LDH)
Elle catalyse la conversion du pyruvate en lactate avec la génération de NAD+
nécessaire pour le maintien da la glycolyse. L'oxamate peut inhiber la LDH mais il
est peu probable que l'inhibition de la LDH seule puisse avoir un effet sur les cellules
de cancer. L'inhibition de la LDH bloque uniquement la production de lactate à partir
du pyruvate ; le pyruvate reste donc disponible pour la fonction mitochondriale dans
les cellules ayant des mitochondries sans défaut. Au contraire, l'inhibition des autres
enzymes en amont dans la voie de la glycolyse comme l'HK ou la PFK, diminue la
glycolyse mais également la quantité du pyruvate disponible pour la génération
mitochondriale d'ATP.
3-5-5. Transkétolases
Elles catalysent les réactions qui permettent à certains métabolites de la voie des
pentoses phosphate de rejoindre la voie glycolytique ou de synthétiser du ribose par
la voie non oxydative des pentoses phosphate à partir des intermédiaires de la
glycolyse. Il a été trouvé qu'une des isoenzymes de la transkétolase (la TKTL1) est
surexprimée dans les tumeurs urothéliales et le cancer du côlon. L'inhibition des
réactions catalysées par la transkétolase réduit la croissance des tumeurs et les
métastases [199]. L'oxythiamine inhibe la transkétolase ainsi que la PDH et supprime
la voie des pentoses phosphate, privant ainsi les cellules de l'intermédiaire GAP
nécessaire pour la génération d'ATP et des substrats (NADPH, ribose-phosphate)
nécessaires pour la synthèse des macromolécules. Cette inhibition est responsable
de l'activité anticancéreuse de l'oxythiamine [200].
3-5-6. Glucose 6-phosphate déshydrogénase (G6PDH)
Il s'agit de l'enzyme qui catalyse la conversion du G6-P en 6-phosphogluconolactone,
la première étape de la voie oxydative des pentoses phosphate. L'inhibition de
l'activité G6PDH diminue la quantité de glucose qui va dans la voie des pentoses
phosphate et prive les cellules des intermédiaires pour la synthèse des nucléotides
et des macromolécules. Le 6-aminonicotinamide inhibe la G6PDH, inhibant ainsi la
voie des pentoses phosphate. L'utilisation clinique du 6-aminonicotinamide est
limitée par ses effets secondaires toxiques, notamment la neurotoxicité [201].
L'imatinib est un inhibiteur de la tyrosine kinase. Il diminue également les activités
G6PDH et HK dans des cellules leucémiques, ce qui conduit à la suppression de la
glycolyse aérobie [202].
3-5-7. Autres facteurs influençant la glycolyse
1- Le développement du glufosfamide représente une stratégie unique profitant de
l'augmentation de la captation du glucose par les cellules cancéreuses. Le
glufosfamide est la conjonction de glucose et d'IFO. Il est capté par le SAAT1, un
transporteur de glucose, surexprimé dans les cellules cancéreuses. Une fois dans la
cellule, le glufosfamide libère l'IFO qui exerce son effet toxique. Le glufosfamide
montre un effet anticancéreux dans des modèles expérimentaux. Il est introduit dans
des essais cliniques pour le traitement des cancers humains du poumon et du
pancréas [203, 204].
2- Dans une étude réalisée par Harmon et al., ils ont obtenu une inhibition de la
captation du glucose et par conséquent, de la prolifération des cellules MCF-7 en
utilisant 10 µM de la naringenine. Il serait possible, selon cette étude, d'atteindre
cette dose physiologiquement. La naringenine peut donc avoir un effet thérapeutique
ou antiprolifératif [205].
3- Tannin-Spitz et al. ont trouvé que les glucosides de cucurbitacine provoquent
l'arrêt du cycle cellulaire et l'apoptose, proposant que ces composés puissent avoir
un effet thérapeutique sur les cellules de cancer du sein MCF-7 [206].
4- L'estradiol stimule la consommation du glucose par des cellules de cancer du sein
en augmentant l'utilisation du glucose par la glycolyse et le cycle des acides
tricarboxyliques [207]. Dans une autre étude, réalisée par Forbes et al. sur des
cellules MCF-7, ils ont trouvé que l'estradiol augmente, de façon significative, le flux
enzymatique dans la voie des pentoses phosphate et la consommation de la
glutamine. Cette glutamine n'est pas oxydée par les mitochondries mais par les voies
de biosynthèse [208].
5- Enfin, Singh et al. ont trouvé que le 2-methoxyestérone, 2-méthoxyestradiol et 2-
méthoxyestérone-O-sulfamide inhibent la captation du glucose par les cellules MCF-
3-6. APPLICATIONS CLINIQUES DE L'EFFET WARBURG
L'essor actuel en cancérologie de l'utilisation in vivo, en tomographie par émission de
positrons (TEP), du fluorodésoxyglucose (FDG) marqué au fluor-18, réactualise la
notion, établie depuis les travaux de Warburg, d'une hyperconsommation de glucose
comme l'une des principales caractéristiques métaboliques des cellules cancéreuses.
Le FDG est un analogue du 2-DOG qui est transporté à l'intérieur de la cellule par
diffusion facilitée assurée par le GLUT-1 (comme le glucose), et est ensuite
phosphorylé par l'HK en FDG-6-P. Celui-ci, n'étant ni métabolisé ni déphosphorylé,
est bloqué dans la cellule et s'y accumule, permettant ainsi la visualisation du tissu
tumoral par la TEP [148]. Pendant les dernières années, l'utilisation de la 18F-FDG
TEP est devenue un moyen très utile en cancérologie pour établir le diagnostic des
tumeurs, localiser les métastases et également pour surveiller la réponse à la
chimiothérapie [210].
3-7. EFFET ANTICANCEREUX DE CAA. DONNEES
Contrairement à son effet néphrotoxique et à sa responsabilité de la toxicité rénale
de l'IFO [43, 80, 113], un effet anticancéreux éventuel du CAA a été très peu étudié.
Il a été démontré que l'IFO possède une activité anticancéreuse plus importante que
le CP dans certaines tumeurs humaines [101] ou expérimentales [211]. L'IFO peut
être efficace dans le traitement des patients prétraités par le CP ou dans les cas
d'échec du traitement par le CP car la résistance croisée entre ces deux isomères
est partielle. La différence structurelle de la position d'un groupe chloroéthyl entre
l'IFO et le CP est à l'origine de la différence du métabolisme entre les deux [96].
Ainsi, le métabolisme de l'IFO produit une quantité plus élevée de CAA que le CP
[95]. Le CAA est considéré comme le responsable de la toxicité rénale rencontré en
cas de traitement par l'IFO.
A part la déplétion du glutathion provoquée par le CAA qui peut faciliter l'action des
agents alkylants, un effet cytotoxique direct du CAA ou une contribution dans
l'efficacité de traitement par l'IFO sont peu étudiés.
1- En 1997, Brüggemann et al. ont publié leurs résultats in vitro sur l'activité
anticancéreuse éventuelle du CAA sur des cellules de cancer du sein (MX1) et du
carcinome de la thyroïde (S117). Ils ont comparé l'effet du CAA et celui du 4-OH-IFO
sur ces cellules, en utilisant des concentrations de CAA proches de celles mesurées
chez 10 patients traités par 5 g/m2 d'IFO (environ 5 µM CAA). Ils ont conclu que le
CAA peut jouer un rôle important dans l'effet antitumoral de l'IFO [108].
2- Börner et al. ont montré que l'administration de CAA à des souris porteuses de
xénogreffes de cancer du sein (cellules MX1) a retardé significativement la
croissance des tumeurs. Le CAA est sept fois moins puissant que le 4-OH-IFO. Ils
ont conclu que la génération de CAA peut expliquer la différence de l'activité
antitumorale et l'absence de la résistance croisée complète entre l'IFO et le CP [212].
La même équipe a publié une étude en 2006 pour élucider les mécanismes d'effet
anticancéreux du CAA sur les cellules de cancer du sein humain (MX1) [213]. Ils ont
démontré que le CAA inhibe la synthèse d'ADN et induit une rupture des brins d'ADN.
Il diminue les concentrations d'ATP par le blocage de la phosphorylation oxydative
des mitochondries. Cette diminution était observée avec des concentrations fortes de
CAA (500 µM) [213].
3- Dans une étude réalisée in vitro sur des cellules de l'adénocarcinome du côlon
(LS147T), Motrescu et al. ont trouvé que 50 µM CAA induit la mort cellulaire et
diminue la consommation d'oxygène d'une façon rapide avec une perte du potentiel
mitochondrial. Les niveaux des molécules oxygénées réactives (ROS) ont augmenté
4- Enfin, Takahashi et al. ont montré que le CAA, à des concentrations supérieures à
30 µM, induit la mort des cellules d'ostéosarcome humain (Saos-2). Les niveaux des
thiols totaux, du glutathion et d'ATP cellulaire ont baissé significativement. Dans
cette étude, la caspase-3 n'était pas active et les cellules ont montré des
caractéristiques de nécrose [215].
III- MATERIELS ET METHODES
1. ETUDE DE LA TOXICITE RENALE DU CAA
1-1. LES MILIEUX D'INCUBATION
1-1-1. Tampon Krebs-Henseleit
Ce tampon est composé de sels minéraux : NaCl (118 mM), KCl (4.7 mM), MgSO4
(1.2 mM), KH2PO4 (1.2 mM), NaHCO3 (25.1.mM) et CaCl2 (2.5 Mm). Il est utilisé pour
la préparation et le recueil des tranches.
1-1-2. Milieu RPMI-1640 sans D-glucose et sans L-cystine
Le milieu RPMI-1640 est un milieu complexe composé des éléments indispensables
pour la survie des cellules : des sels inorganiques, des acides aminés, des vitamines
etc. (Tableau 1). Dans nos expériences, ce milieu est sans L-glutamine, sans L-
cystine et sans D-glucose.
1-2. LES ANIMAUX
Les expériences sont réalisées sur des rats Wistar mâles de 280-350 g (âgés de 9-
11 semaines) fournis par le laboratoire Charles River, France, ayant accès libre à
l'eau et à la nourriture selon un régime standard. Toutes les expériences ont été
réalisées en respectant un protocole approuvé par l'Université Claude Bernard,
Tableau 1 : Composition du milieu RPMI-1640
Composant
Quantité
Composant
Quantité
Sels Inorganiques
Acides Aminés
Vitamines
L-hydroxy-proline
D-Ca-panthothénate
Chlorure de Choline
L-phénylalanine
Acide p-aminobenzoique
Autres composants
Glutathion (réduit)
Rouge de phénol



1-3. TECHNIQUE DES TRANCHES DE CORTEX RENAL DE RAT
1-3-1. Réalisation des tranches de cortex rénal
Les rats sont anesthésiés par une injection intra péritonéale de 35 mg/kg de poids de
pentobarbital de sodium (SANOFI, France). Les deux reins sont immédiatement
prélevés, via une laparotomie, et plongés dans du tampon glacé de Krebs-Henseleit.
Ils sont ensuite décapsulés et coupés en cylindres (carottes) de 5 mm de diamètre à
l'aide d'un emporte-pièce monté sur une perceuse.
Les tranches, d'épaisseur calibrée (200-250 µm), sont obtenues à l'aide d'un
microtome de Krumdieck (Figure 10-1).
Figure 10-1 : Le microtome de Krumdieck

La vitesse de coupe est réglée à 50 cycles par minute. L'épaisseur des tranches est
déterminée à l'aide d'un poids et d'une vis micrométrique.
Le bloc de microtome contient du tampon Krebs-Henseleit froid et gazé avec un
mélange d'O2 (95%) et CO2 (5%). La carotte est placée dans un puits de 5 mm de
diamètre puis elle est coupée en tranches grâce à des mouvements de va-et-vient
par une lame (Figure 10-2).
Figure 10-2 : Le microtome de Krumdieck. A- lame, B- puits contenant la carotte de tissu, C-
sens du mouvement du tampon créé par une pompe, D- réservoir pour la collection des
tranches, E- vis micrométrique, F- valve. D'après Parrish et al. Life Sciences, Vol. 57, No. 21,
pp. 1887-1901, 1995.
Les tranches sont recueillies à la sortie du réservoir du microtome dans du tampon
Krebs-Henseleit et maintenues au froid jusqu'à leur incubation. Les tranches de la
médullaire sont éliminées et nous conservons uniquement les tranches de cortex.
1-3-2. Mise en roller et incubation
Figure 11 : Un roller
Les tranches sont déposées sur la grille d'un
roller (une tranche par roller). Ce dernier est un
petit élément cylindrique en téflon composé
d'une grille en acier inoxydable (Figure 11). Cet
élément est placé dans un flacon à scintillation
contenant 2 ml de milieu d'incubation et fermé
par un bouchon troué en son centre permettant

Certain flacons ne contiennent pas de tranches ; ils serviront de temps zéro et seront
pris comme référence lors des dosages enzymatiques.
Le milieu utilisé est un milieu RPMI-1640 sans D-glucose et sans L-cystine, qui
contient du L-lactate à une concentration physiologique de 1 mM. Un antibiotique, (la
gentamycine 0.05 mg/ml), est ajouté au milieu pour les incubations de 8 heures ou
plus. Le CAA est ajouté au milieu à une concentration finale variable entre 25 et 75
µM. L'incubation se déroule dans un incubateur multi-gaz (HERAEUS, HERAcell 150)
saturé en humidité à 37 °C, en présence de 40% d'O2 et 5% de CO2.
Figure 12 : Agitateur STOVALL Low Profile Roller
Les flacons contenant les rollers
sont positionnés sur un agitateur
rotatif (STOVALL Low Profile Roller)
(Figure 12) qui tourne à une vitesse
de 5 tours par minute. Ce système
d'agitation permet aux tranches
d'être alternativement en contact
avec le milieu d'incubation et avec
le mélange gazeux.
En fin d'incubation, le milieu d'incubation est recueilli pour effectuer les différents
dosages. Il est acidifié par l'acide perchlorique (HClO4) (2% v/v, concentration finale),
neutralisé avec un mélange de KOH (20%) et H3PO4 (1%), puis lyophilisé. La
lyophilisation est effectuée dans un Speed Vac Concentrator (SAVANT) afin de
concentrer le milieu et de rendre les métabolites détectables par les méthodes de
dosage enzymatique. Le milieu est concentré 10. Contrairement au lactate, existant
en concentrations importantes dans le milieu d'incubation, et au pyruvate, étant
fragile à la lyophilisation, le glucose est dosé après la lyophilisation.
Les tranches sont recueillies pour effectuer d'autres dosages. Elles sont mises dans
un volume de 0.2 ml d'acide perchlorique (7%, v/v, concentration finale) et agitées
sur un vortex, ce qui permet de casser les membranes cellulaires. Une centrifugation
à 4000g pendant 5 minutes à 4 °C permet de récupérer le surnageant. Ce dernier est
neutralisé avec un mélange de KOH (20%) et H3PO4 (1%) et ensuite utilisé pour
doser l'ATP, le glutathion et les thiols totaux. Le culot résultant de la centrifugation
sera dissous dans de la soude (NaOH 0.1 N) pour doser le contenu protéique des
Dans les expériences réalisées pour étudier les différentes activités enzymatiques,
les tranches, en fin d'incubation, sont homogénéisées dans des tampons spécifiques
selon l'activité étudiée. Dans toutes les expériences, des tranches non incubées sont
traitées de la même façon que les tranches incubées. Elles serviront de témoins
"temps zéro". Les conditions expérimentales sont réalisées en double ou en triple.
1-3-3. Etude de la radioactivité
Dans les expériences où un produit marqué ([U-14C]-lactate) est utilisé, l'incubation
des tranches de cortex rénal de rat est réalisée dans des Erlenmeyer de 25 ml
possédant un puits central et placés dans un bain marie oscillant à 90 cycles par
minutes à 37 °C. Les fioles sont gazées pendant une minute par un mélange de 95%
O2 et 5% CO2 puis fermées par un bouchon à jupe de manière à éviter le
dégagement de CO2 dans l'atmosphère. L'incubation s'effectue pendant 4 heures
dans le milieu RPMI-1640 (sans D-glucose et sans L-cystine) qui contient 1 mM de
[U-14C]-lactate comme substrat en présence ou en absence de 75 µM CAA. L'acide
perchlorique est introduit en fin d'incubation via une seringue et une aiguille longue à
travers le bouchon. De la même façon, 1 ml de soude 1 N est introduit dans le puits
central pour capter le [14CO2]. Les fioles sont agitées pendant 2 heures environ à
température ambiante. Ensuite, les milieux sont traités comme indiqué
précédemment et le contenu du puits central est prélevé pour la mesure de la
formation de [14CO2]. Dans toutes les expériences, les conditions expérimentales
sont réalisées en double.
1-4. METHODES ET TECHNIQUES EXPERIMENTALES
1-4-1. Dosage des métabolites
Les dosages sont réalisés dans des microplaques de 96 puits à fond plat (GRENIER)
et les mesures sont effectuées dans un spectrophotomètre lecteur de plaque
SPECTRAMAX 340PC (MOLECULAR DEVICES, St Grégoire, France). La longueur d'onde
est réglée à 340 nm pour tous les dosages (sauf les protéines, à 590 nm, et les thiols,
Ces dosages sont basés sur la différence d'absorption entre NAD+ et NADH ou entre
NADP+ et NADPH. La quantité en mol/l du coenzyme réduit ou oxydé qui apparaît
durant la réaction est égale à la quantité de produit présent dans le milieu. La
quantité du produit est déterminée par calcul à partir de la variation de densité
optique (∆DO).
- Dosage d'ATP
Il est effectué selon la méthode de Trautschold, Lamprecht et Schweitzer [216] :
Glucose + ATP Glucose-6-phosphate + ADP
Glucose-6-phosphate + NADP+ 6-phosphogluconolactone + NADPH + H+
Le tampon de dosage est du Tris-HCl 0.5 M à pH 7.4 additionné de glucose et de
MgCl2. La G6PDH est ajoutée dans le tampon et l'HK démarre la réaction. La
quantité de NADPH formée est mesurée.
- Dosage de glucose
Il est effectué selon la méthode décrite par Kunst, Draeger et Ziegenhorn [217] :
Glucose + ATP Glucose-6-phosphate + ADP
Glucose-6-phosphate + NADP+ 6-phosphogluconolactone + NADPH + H+
Le tampon de dosage est du Tris-HCl 0.1 M à pH 8 en présence d'ATP et de MgCl2.
Le NADPH formé correspond au glucose présent.
- Dosage de glutathion
Les concentrations de glutathion total (réduit (GSH) et oxydé (GSSG)) sont
mesurées par la méthode de Griffith [218] :
DTNB + NADPH + H+ 2TNB + NADP+
La détermination des dérivés sulphydriles dans un milieu sans protéines à l'aide du
réactif d'Ellman (DTNB, 5,5'-dithiobis-2-nitrobenzoate) permet une détermination
rapide des concentrations de glutathion total. La méthode enzymatique avec la
glutathion réductase (GR) est une méthode simple, sensible et spécifique.
Le glutathion réagit non enzymatiquement avec le DTNB et conduit à la formation de
GSSG. Les concentrations de DTNB, NADPH et GR sont choisies en fonction de la
concentration de glutathion total à mesurer. Une réaction de recyclage de glutathion
à l'aide de la GR est utilisée pour permettre le dosage des quantités très faibles de
glutathion total (limite de sensibilité 0.1 µM) [218]. Le taux de formation de TNB (5-
thio-2-nitrobenzoic acide) est déterminé au spectrophotomètre à 405 nm (coloration
jaune) et calibré par une gamme étalon de standard.
- Dosage de lactate
Il s'effectue selon la méthode de Gutmann et Wahlefeld [219] :
Lactate + NAD+ Pyruvate + NADH + H+
Le tampon est de la glycine 0.5 M à pH 9.5 additionné d'hydrazine pour déplacer la
réaction vers la droite et piéger le pyruvate sous forme de pyruvate hydrazone.
L'enzyme de la réaction est la LDH. La formation de NADH est mesurée.
- Dosage des protéines
Les tranches sont dissoutes dans la soude (NaOH 0.1 N). Le dosage s'effectue selon
la méthode de Lowry [220], une méthode colorimétrique, à une longueur d'onde de
590 nm. Le tampon est composé de carbonate disodique dissous dans de la soude
0.1 N, de tartrate de sodium et potassium et de sulfate de cuivre. La lecture se fait en
point final par comparaison à une gamme étalon de sérum albumine bovine (BSA).
- Dosage de pyruvate
Le pyruvate est dosé selon la méthode de Lamprecht et Heinz [221] :
Pyruvate + NADH + H+ Lactate + NAD+
Le milieu de dosage est le tampon de Soerensen, (tampon phosphate :
K2HPO4/KH2PO4 0.1 M) à pH 7. La décroissance de NADH est mesurée.
- Dosage des thiols totaux
Le dosage de thiols totaux est effectué sur les tranches. En fin d'incubation, les
tranches sont homogénéisées dans l'acide perchlorique 7% puis centrifugées
pendant 5 minutes à 12000g.
Le dosage est effectué sans neutraliser le milieu selon la méthode d'Ellman [222]
basée sur la réaction des thiols avec le DTNB pour former le TNB. Il se déroule dans
un tampon phosphate de sodium 0.25 M à pH 7.4 avec 0.3 mM de DTNB. La lecture
au spectrophotomètre se fait à 412 nm contre un blanc d'eau. La valeur de (ε = 13.6
x 103 M-1 cm-1) est utilisée pour les calculs.
1-4-2. Dosage des activités enzymatiques
Les dosages des activités enzymatiques sont réalisés sur des tranches de cortex
rénal incubées 4 heures à 37 °C dans le milieu RMPI (sans D-glucose et sans L-
cystine) contenant 1 mM L-lactate, en présence ou en absence de 75 µM CAA.
En fin d'incubation, les tranches sont homogénéisées dans un tampon d'extraction
(variable selon l'activité mesurée). Un échantillon d'homogénat est prélevé dans la
soude 0.1 N pour le dosage des protéines, puis l'homogénat est centrifugé à 4 °C
pour obtenir un surnageant sur lequel le dosage est effectué.
- Dosage de GAPDH
Dix tranches sont homogénéisées dans 1 ml (0.1 ml par tranche) de tampon
d'extraction (un tampon TEA-HCl 10 mM à pH 7.6, additionné de KCl 100 mM et
d'EDTA 0.5 mM). Les échantillons sont centrifugés pendant 20 minutes à 16000g.
Le dosage est effectué dans un tampon TEA-HCl 100 mM à pH 7.6, additionné
d'EDTA 0.5 mM, de NADH 0.2 mM, d'ATP 1 mM, de MgSO4 2 mM, de
phosphoglycérate kinase (PGK), et de 3-PG 6 mM ; selon la méthode de Boehringer
décrite par Sakai et al. [223] :
3-P-Glycérate + ATP 1.3-diphosphoglycerate + ADP
1.3-diphosphoglycerate + NADH + H+ Glycéraldéhyde-3-P + NAD+ + Pi
L'ajout de la GAPDH, contenue dans l'échantillon, déplace la deuxième réaction vers
la droite. La décroissance de NADH est mesurée, en cinétique pendant 5 minutes à
Une série d'échantillons avec le tampon sans 3-PG est réalisée lors de dosage. Elle
sert de blanc qui sera éventuellement soustrait de la valeur de GAPDH
- Dosage de G6Pase
Six tranches sont homogénéisées dans 0.6 ml (0.1 ml par tranche) de tampon
d'extraction (Imidazole 50 mM à pH 7.2, EDTA 1 mM et BSA 0.02%). Les
homogénats sont centrifugés pendant 5 minutes à 7000g.
Le dosage de cette activité comporte deux étapes [224]. La première est la synthèse
du glucose par l'intermédiaire de la G6Pase :
Glucose-6-phosphate Glucose + Pi
Elle s'effectue dans un tampon Imidazole-HCl 200 mM à pH 6.7 additionné d'EDTA 2
mM et de BSA 0.05%. Elle se fait dans un bain marie à 37 °C pendant une heure.
Une série d'échantillons sans substrat (G6-P) sert de blanc. La réaction est arrêtée
par l'ébullition pendant 2-3 minutes.
La deuxième étape est la quantification du glucose formé :
Glucose + ATP Glucose-6-phosphate + ADP
ADP + Phosphoénolpyruvate Pyruvate + ATP
Pyruvate + NADH Lactate + NAD+
Elle s'effectue dans un tampon Imidazole-HCl 75 mM à pH 7.2 additionné de KCl 112
mM, d'ATP 0.17 mM, de PEP 0.5 mM, de MgCl2 3.3 mM, de β mercaptoéthanol 3.3
mM, de NADH 0.12 mM, de PK 1.67 U/ml et de LDH 10 U/ml. La réaction est
démarrée par l'ajout d'HK 15 U/ml. La décroissance de NADH est mesurée.
- Dosage de F1,6-BPase
Le tampon d'extraction de cette enzyme et du Tris-HCl 0.02 M à pH 7.8 (0.2 ml de
tampon par tranche). L'homogénat est centrifugé pendant 5 minutes à 7000g.
Le dosage s'effectue selon la méthode de Latzko et Gibbs [225] :
Fructose-1,6-bisphosphate Fructose-6-phosphate + Pi
Fructose-6-phosphate Glucose-6-phosphate
Glucose-6-phosphate + NADP 6-Phosphogluconate + NADPH
Le dosage s'effectue dans un tampon Imidazole-HCl 50 mM pH7, additionné de BSA
0.05%, de β mercaptoéthanol 2 mM, de MgCl 2 2 mM, de NADP 0.2 mM, de G6PDH
0.25 U/ml, de phosphoglucoisomérase (PGI) 0.35 U/ml et de F1,6-P2 0.1 mM. Une
série d'échantillons sans ce dernier sert de blanc au dosage. Celui-ci est réalisé en
cinétique pendant 5 minutes à 25 °C. Le NADPH formé est mesuré.
- Dosage de PEPCK
Le tampon d'extraction est le même que pour la GAPDH. L'homogénat est centrifugé
pendant 5 minutes à 7000g.
Deux étapes sont nécessaires pour doser l'activité PEPCK [226]. La première est la
transformation d'oxaloacétate en PEP :
Oxaloacétate + ITP PEP + IDP + CO2
Elle s'effectue pendant une heure dans un bain-marie à 37 °C dans un tampon
Imidazole sulfate 75 mM pH 7.2 additionné d'inosine triphosphate (ITP) 6 mM, de
MgSO4 12 mM, de MnSO4 0.36 mM, d'EGTA 0.12 mM, de β mercapto
mM, de BSA 0.05% et d'oxaloacétate 3 mM. Une série d'échantillons sans
oxaloacétate représente le blanc de dosage. La réaction est arrêtée par l'ébullition
pendant 2-3 minutes (qui détruit l'oxaloacétate en excès). Dix µl d'eau oxygénée 3%
sont ajoutés pendant 2-3 minutes à l'ébullition pour détruire le pyruvate (provenant
de l'hydrolyse spontanée de 5-10% d'oxaloacétate à 37 °C). L' H2O2 est éliminée par
l'ajout de 10 µl de catalase (10 minutes d'attente à température ambiante).
La deuxième étape est le dosage de PEP formé :
PEP + ADP Pyruvate + ATP
Pyruvate + NADH Lactate + NAD+
Elle s'effectue dans un tampon Imidazole-HCl 100 mM à pH 7.2 additionné de KCl
120 mM, d'ADP 0.3 mM, de NADH 0.3 mM et de LDH 10 U/ml. L'ajout de PK
démarre la réaction. La décroissance de NADH est mesurée.
- Dosage de LDH
Cette activité est dosée sur les mêmes échantillons préparés pour le dosage de
l'activité G6Pase (tampon d'extraction : Imidazole 50 mM à pH 7.2, EDTA 1 mM et
BSA 0.02%). Ce dosage s'effectue en cinétique à 340 nm pendant 5 minutes à 25 °C
dans un tampon phosphate 0.1 M (K2HPO4 / KH2PO4) à pH 7.5, et selon la méthode
de Vassault [227] :
Pyruvate + NADH + H+ Lactate + NAD+
La décroissance de l'absorbance de NADH est mesurée.
1-5. MESURES RADIOACTIVES
Le carbone 14 est un émetteur
β -. Ces particules β- entrent en collision avec les
molécules du liquide scintillant et les excitent. Ceci induit l'émission de photons qui
seront détectés par la cellule photomultiplicatrice du compteur puis transformés sous
forme d'impulsions électriques. Ceci conduira à la détermination du nombre de coups
par minute (cpm). Le signal électrique est proportionnel à la quantité de photons, et
par conséquent, à la radioactivité.
Pour déterminer la radioactivité, le contenu du puits central est récupéré puis ajouté
à 2.5 ml d'un liquide scintillant (
ULTIMA GOLD , PACKARD) dans des fioles à scintillation.
0.1 ml supplémentaire de soude 1 N est ajouté au liquide scintillant pour le rendre
alcalin et éviter le dégagement de CO2 lors de l'ajout du contenu du puits central. Les
comptages sont aussi réalisés sur un aliquote de milieu non neutralisé au temps zéro
de l'expérience. Ceci permet de mesurer la radioactivité spécifique du lactate marqué,
ajouté au début d'incubation. Les comptages sont réalisés sur 2 cycles de 2 minutes
dans le compteur à scintillation liquide (PACKARD, Tri-Carb 2100TR).
1-6. ETUDE DES MITOCHONDRIES ISOLEES
Cette étude est réalisée sur des mitochondries isolées de tranches de cortex rénal
de rat préalablement incubées pendant 4 heures dans le milieu RPMI-1640 (sans D-
glucose et sans L-cystine) contenant 1 mM de L-lactate, en présence ou en absence
de 75 µM CAA. L'isolation des mitochondries s'effectue selon la méthode décrite par
Cain et Skilleter [228], dans un tampon d'extraction (Tris-HCl 10 mM à pH 7.4,
sucrose 0.25 M et EGTA 1 mM).
Dans nos expériences, 25 ou 30 tranches sont homogénéisées au Potter dans un
volume de 1.5 ml de tampon d'extraction. L'homogénat est ensuite centrifugé
pendant 10 minutes à 460g à 4 °C. Le surnageant est récupéré et centrifugé pendant
7 minutes à 12500 g. Le culot, qui correspond à la fraction mitochondriale, est repris
dans 100 µl de tampon d'extraction (ce qui donne une concentration d'environ 20 mg
de protéines par 100 µl de tampon).
La respiration des mitochondries (la consommation d'O2) est mesurée par une
électrode à oxygène type Clarck dans un tampon de respiration (Tris-HCl 10 mM à
pH 7.4, sucrose 0.25 M, KH2PO4 5 mM, KCl 10 mM, MgCl2 5 mM et EGTA 1 mM) à
30 °C. L'électrode est connectée à un enregistreur. La consommation d'O2 est
mesurée par l'entrée des électrons au niveau du complexe I et du complexe II de la
chaîne respiratoire.
FADH2 + ½ O2 FAD + H2O
Le mélange malate + glutamate et le mélange malate + pyruvate sont des donneurs
d'électrons à la chaîne respiratoire au niveau de complexe I. Le succinate est un
donneur d'électrons au niveau de complexe II. Après avoir ajouté chacun de ces
donneurs, l'ADP (20 mM) est ajouté pour étudier le couplage (oxydation /
phosphorylation). La consommation d'O2 est ramenée à la quantité des protéines et
à l'unité de temps.
1-7. CALCULS ET EXPRESSION DES RESULTATS
1-7-1. Dosages et activités enzymatiques
Les substrats et métabolites sont dosés par des méthodes enzymatiques. La
concentration d'une substance est déterminée par la loi de Beer-Lambert :
DO = ε . l . c
DO : densité optique d'échantillon à 340 nm.
ε : coefficient d'absorption de NADH ou NADPH à 340 nm (= 6.22 x 103 M-1 cm-1).
l : longueur du trajet optique.
c : concentration du coenzyme dans l'échantillon (mol . L-1).
Deux lectures de la densité optique (DO) sont réalisées pour les dosages en point
final avant et après l'ajout de l'enzyme qui catalyse la réaction de dosage. Une
différence de DO (
ΔDO) est ainsi déterminée pour chaque échantillon. Elle est
corrigée par la (ΔDO) d'un blanc d'eau ultra pure traité dans les mêmes conditions.
La lecture en cinétique s'effectue pendant 5 minutes. L'ajout de l'enzyme lance la
cinétique. La valeur obtenue de DO est aussi corrigée par un blanc.
La concentration des différents métabolites est déterminée indirectement par la
quantification de la croissance ou de la décroissance du coenzyme (NADH – NAD+,
ou NADPH – NADP+).
La consommation ou la production des métabolites est déterminée par la différence
entre la valeur mesurée après l'incubation et le "temps zéro". Les valeurs obtenues
exprimées en micromoles par ml de milieu sont ensuite ramenées à la quantité des
protéines correspondant à la condition expérimentale. Les résultats sont donc
exprimés en micromoles de substrat par mg de protéines.
1-7-2. Comptages radioactifs
Les résultats de comptage sont exprimés en coups par minute (cpm). La radioactivité
spécifique de substrat utilisé ([U-14C]-lactate) est d'abord calculée en divisant la
radioactivité totale présente dans le milieu au "temps zéro" par le nombre de
micromoles de substrat présent dans le milieu au même temps. L'activité spécifique,
exprimée en cpm/µmol, est utilisée comme facteur de conversion pour transformer
les cpm, obtenus par le comptage, en micromoles de [14CO2] libéré dans
l'atmosphère de la fiole pendant le temps d'incubation. La quantité de [14CO2]
correspond à la quantité de lactate oxydé.
1-8. ANALYSES STATISTIQUES
Les résultats sont exprimés en moyenne ± erreur standard à la moyenne (SEM).
Une analyse de variance (ANOVA) est appliquée pour des mesures répétées suivie
d'un test de Fisher pour comparer les conditions d'incubation au cours du temps
ainsi que les valeurs obtenues après l'incubation de tissu sans et avec CAA, dans
toutes les séries expérimentales.
Les analyses statistiques sont effectuées à l'aide d'un logiciel (STATVIEW). Des
probabilités inférieures à 0.05 sont considérées comme statistiquement significatives.
1-9. PRODUITS ET REACTIFS
Le CAA, est fourni par Sigma-Aldrich, Germany.
Le milieu RPMI-1640 est fourni par le laboratoire Eurobio, France.
Le [13C] lactate est fourni par Cambridge Isotope Laboratories, Inc, USA.
La [2-13C] glycine est fournie par Euriso-top France.
Les enzymes, coenzymes et le L-lactate sont fournis par Roche, France.
Les sels minéraux sont fournis par MERCK, Germany et par Sigma-Aldrich, Germany.
2. ETUDE DE L'EFFET DU CAA SUR LES CELLULES MCF-7
2-1. MILIEUX ET MATERIELS DE CULTURE
La culture des cellules de cancer du sein (MCF-7), provenant à l'origine de l'ATCC
(American Type Culture Collection, Manassas, VA, USA) est réalisée dans un milieu
RPMI-1640 contenant 2 g/l de glucose. Du sérum de veau fœtal (SVF) est ajouté à
une concentration de 10% pour démarrer la culture à partir des cellules congelées et
de 5% pour entretenir les cellules et réaliser l'étude de l'effet du CAA. Deux substrats
sont également ajoutés au milieu : la glutamine (2 mM, concentration finale) et le
pyruvate (1 mM, concentration finale). Pour éviter tout risque de contamination, des
antibiotiques (un mélange de 100 UI/ml de pénicilline et 100 µg/ml de streptomycine)
et un antifongique (amphotéricine B, 1.25 µg/ml) sont également ajoutés au milieu.
Tous les matériels utilisés sont stériles. La culture s'effectue dans des boîtes de pétri
standard (surface 58,95 cm²) ou dans des plaques à 6 puits dont la surface de puits
est de 9.6 cm². La culture a lieu dans un incubateur monogaz (HERAEUS, HERAcell
150) humidifié à 37 °C en présence de 5% CO2. Les expériences réalisées en
hypoxie ont lieu dans un incubateur multigaz en présence de 5% CO2 et 5% O2 (en
remplaçant l'oxygène par l'azote). Toute manipulation des matériels et des cellules
est réalisée d'une façon stérile sous un poste de sécurité microbiologique (PSM).
2-2. DECONGELATION DES CELLULES
Pour lancer une culture, les cellules, déjà conservées dans des cryotubes dans
l'azote liquide, doivent être décongelées. Les tubes sont décongelés dans un bain
marie à 37 °C. Leur contenu est ensuite repris dans 15 ml de milieu RPMI-1640
contenant 10% SVF et centrifugé pendant 5 minutes à 1200 rpm. Le surnageant (le
milieu) est éliminé, éliminant ainsi le DMSO (diméthylsulfoxide) qui avait été ajouté
lors de la congélation des cellules. Le culot de cellules est repris dans 3 ml de milieu
contenant 10% SVF pour obtenir une suspension des cellules. Cette dernière est
mise dans une boîte de Pétri contenant 9 ml de milieu (SVF 10 %). Le volume final
obtenu est 12 ml par boîte. La boîte est ensuite introduite dans l'incubateur.
Les cellules commencent à adhérer au fond des boîtes après 12-18 heures. Elles
croissent en monocouche et, 48 heures plus tard, elles atteignent 80-90% de
confluence. Elles peuvent donc être manipulées et transférées soit dans des plaques
à 6 puits pour réaliser une étude sur l'effet du CAA, soit dans d'autres boîtes de Pétri
pour obtenir une nouvelle génération de cellules.
2-3. TRYPSINATION DES CLLULES
Afin de décoller les cellules du fond des boîtes, de la trypsine-EDTA 0.25% (qui
contient 2.5 g de trypsine porcine et 0.2 g d'EDTA-4Na par litre) est utilisée.
Le milieu d'incubation, qui contient les cellules mortes et les débris cellulaires, est
éliminé à l'aide d'une pompe à vide. La couche cellulaire est rincée deux fois par 3-4
ml environ de PBS (phosphate-buffered saline) qui est également éliminé. La
trypsine-EDTA (1.5 ml, diluée au ½ dans du PBS) est appliqué sur les cellules et agit
pendant 2 minutes à 37 °C. Il faut éventuellement taper légèrement sur la boîte pour
détacher les cellules. Une fois les cellules détachées (ceci est vérifié à l'oeil), 4.5 ml
de milieu RPMI-1640 contenant 5% SVF sont ajoutés afin de désactiver la trypsine.
Le volume final de la suspension est de 6 ml. A l'aide d'une pipette, les cellules sont
décollées par plusieurs jets ce qui permet également d'homogénéiser la suspension
Deux boîtes de Pétri sont préparées en mettant 9 ml de milieu RPMI-1640 avec 5%
SVF par boîte, puis 3 ml de la suspension de cellules sont ajoutés par boîte pour
obtenir un volume final de 12 ml.
2-4. CONGELATION DES CELLULES
Pour procéder à la congélation des cellules, celles-ci sont trypsinées comme décrit
dans le paragraphe précédent. Il est préférable de congeler les cellules en phase de
croissance avant d'atteindre la confluence.
La suspension de cellules est reprise dans un tube et centrifugée pendant 5 minutes
à 1200 rpm. Le surnageant est ensuite éliminé, et le culot est repris dans le milieu
RPMI-1640 sans SVF. Le DMSO (dilué au ½ dans le RPMI) est ajouté à la
suspension pour obtenir un mélange de 4 volumes de la suspension des cellules et
un volume de DMSO. La concentration finale de DMSO est 10%.
La suspension est mise dans un cryotube et congelée progressivement : d'abord
dans de la glace, puis au congélateur à -20 °C, ensuite au congélateur à -80 °C et en
fin dans l'azote liquide.
2-5. ETUDE DE L'EFFET DU CAA
Pour réaliser l'étude de la toxicité du CAA, et après avoir obtenu la suspension de
cellules comme décrit ci-dessus, les cellules sont ensemencées dans des plaques à
6 puits à une densité de 6x105 cellules par puits dans un volume final de 2 ml de
milieu RPMI-1640 contenant 5% SVF. Cette densité a été maintenue pour toutes les
expériences réalisées dans cette étude.
Toutes les expériences sont réalisées 48 heures après l'ensemencement des
cellules quand elles sont en pleine phase de croissance (80-90% de confluence). A
ce stade, l'effet du CAA est étudié. L'effet de plusieurs concentrations de CAA (0, 5,
10, 25, et 50 µM) est étudié pendant 24 heures d'incubation. Les conditions
expérimentales sont réalisées en triple.
2-6. DOSAGES ENZYMATIQUES
En fin d'incubation, les échantillons sont recueillis pour réaliser les différents dosages.
Les dosages sont réalisés dans des microplaques de 96 puits à fond plat et les
mesures sont effectuées dans un spectrophotomètre lecteur SPECTRAMAX 340 PC
(MOLECULAR DEVICES, St Grégoire, France).
Les valeurs obtenues pour les différents dosages sont rapportées au nombre de
cellules correspondant, déterminé dans des puits parallèles. Pour déterminer la
viabilité et le nombre total de cellules après l'incubation, elles ont été rincées deux
fois par le PBS, puis décollées des fonds de puits en utilisant de la trypsine-EDTA.
Après avoir incubé les cellules pendant 5 minutes avec l'iodure de propidium (2
µg/ml), elles ont été analysées dans un cytomètre (
PARTEC, GALAXY PRO , Germany).
Les calculs et l'expression des résultats sont décrits précédemment (page 68).
2-6-1. Dosage de glucose et de lactate
Pour le dosage de ces métabolites, un prélèvement du contenu des puits est réalisé.
Le milieu est ensuite acidifié par HClO4 (2% v/v, concentration finale) puis neutralisé
avec un mélange de KOH (20%) et H3PO4 (1%). Le dosage de glucose est effectué
selon la méthode décrite par Kunst, Draeger et Ziegenhorn [217] et celui de lactate
est réalisé selon la méthode décrite par Gutmann et Wahlefeld [219]. Ces méthodes
sont détaillées précédemment (pages 61-62).
2-6-2. Dosage d'ATP
Après avoir éliminé tout le milieu d'incubation, l'HClO4 7% est ajouté directement sur
les cellules. La plaque est agitée sur un agitateur de plaques. Ensuite, l'acide est
récupéré et neutralisé avec KOH (20%) et H3PO4 (1%). Le dosage d'ATP s'effectue
selon la méthode de Trautschold, Lamprecht et Schweitzer [216] (page 61).
2-6-3. Dosage des activités enzymatiques
Les activités de certaines enzymes de la glycolyse sont mesurées dans les cellules
incubées pendant 24 heures dans le milieu RPMI-1640 contenant 5% de SVF et en
présence de différentes concentrations de CAA. En fin d'incubation, le milieu est
éliminé, puis le tampon d'extraction des enzymes est ajouté directement sur les
cellules. C'est un tampon TEA-HCL 10 mM à pH 7.6 additionné de KCl 100 mM,
d'EDTA 0.5 mM et de Triton-X100 2 mg/ml. Les plaques sont agitées sur un agitateur
de plaques pendant quelques minutes. Une demi-heure plus tard, le tampon est
récupéré et les dosages des activités HK, G6PDH, PFK, GAPDH et PK sont effectués.
- Dosage d'HK
Ce dosage s'effectue selon la méthode décrite par Bergmeyer, Gawehn et Grassl
[229], selon la réaction suivante :
D-Glucose + ATP D-Glucose-6-P + ADP
D-Glucose-6-P + NADP+ D-Gluconate-6-P + NADPH + H+
Le tampon de dosage est composé de TEA 40 mM à pH 7.6 additionné de glucose
222 mM, de MgCl2 8 mM, de NADP 0.91 mM, d'ATP 0.64 mM et de G6PDH 0.55
U/ml. La croissance de NADPH est mesurée en cinétique (340 nm) pendant 5
minutes à 25 °C.
- Dosage de G6PDH
Il s'effectue selon la méthode décrite par Bergmeyer, Gawehn et Grassl [229] :
D-Glucose-6-P + NADP+ D-Gluconate-6-P+ NADPH + H+
Le tampon de dosage est constitué de TEA 86 mM à pH 7.6 additionné de MgCl2 6.9
mM, de G6-P 1 mM et de NADP 0.39 mM. La croissance de NADPH est mesurée en
cinétique (340 nm) pendant 5 minutes à 25 °C.
- Dosage de PFK
Le dosage se fait selon la méthode décrite par Bergmeyer, Gawehn et Grassl [229] :
Fructose-6-P + ATP Fructose-1,6-P2 + ADP
ADP + PEP ATP+ Pyruvate
Pyruvate + NADH + H+ L-Lactate + NAD+
Le dosage s'effectue dans un tampon Tris 70 mM à pH 8.5 additionné de MgSO4 1.4
mM, de KCl 4.5 mM, de PEP 0.71 mM, de F1,6-P2 0.64 mM, de F6-P 1.8 mM, d'ATP
1.1 mM, de NADH 0.4 mM, de PK 4.2 U/ml et de LDH 9.6 U/ml. La décroissance de
NADH est mesurée en cinétique pendant 5 minutes (25 °C, 340 nm).
- Dosage de GAPDH
Le dosage est effectué dans un tampon de TEA-HCl 100 mM à pH 7.6 additionné
d'EDTA 0.5 mM, de NADH 0.2 mM, d'ATP 1 mM, de MgSO4 2 mM, de PGK 50 µl, et
de 3-PG 6 mM ; selon la méthode de Boehringer décrite par Sakai et al. [223] :
3-P-Glycérate + ATP 1.3-diphosphoglycerate + ADP
1.3-diphosphoglycerate + NADH + H+ Glycéraldéhyde-3-P + NAD+ + Pi
L'ajout de la GAPDH déplace la deuxième réaction vers la droite. La décroissance de
NADH est mesurée en cinétique pendant 5 minutes à 25 °C.
- Dosage de PK
Il s'effectue selon la méthode décrite par Gutmann et Bernt [230] :
PEP + ADP Pyruvate+ ATP
Pyruvate + NADH + H+ L-Lactate + NAD+
Le dosage s'effectue dans un tampon TEA-HCl 123 mM à pH 7.5 additionné de KCl
92.5 mM, de MgSO4 16.2 mM, d'un mélange de NADH 185 µM et PEP 1 mM, et de
LDH 2.5 U/ml. L'ajout d'ADP 3 mM déclenche la réaction. La décroissance de NADH
est mesurée en cinétique (25 °C ; 340 nm).
2-6-4. Dosage des métabolites de la voie de la glycolyse
Parmi les métabolites de la voie de la glycolyse, nous avons dosé le F1,6-P2, le
DHAP et le GAP intracellulaires. Le dosage est réalisé sur les mêmes échantillons
préparés pour le dosage des activités enzymatiques. C'est un dosage unique qui
permet de doser les trois métabolites. Il est décrit par Michal et Beutler [231] selon
les réactions suivantes :
Fructose-1,6-P2 DHAP+ GAP
DHAP + NADH + H+ L-Glycérol-3-P + NAD+
Dans une première étape, l'échantillon est mélangé avec du tampon TEA-HCl 200
mM à pH 7.6 et du NADH (17 µM, concentration finale) puis la glycérol-3-phosphate
déshydrogénase (GDH) est ajoutée (0.13 U/ml, concentration finale). La première
lecture correspond à la quantité de DHAP trouvé dans l'échantillon. Dans la
deuxième étape, la triose phosphate isomérase (TIM) est ajoutée (0.83 U/ml,
concentration finale). La deuxième lecture correspond à la quantité de GAP. Enfin,
en ajoutant l'aldolase (0.045 U/ml, concentration finale) la troisième lecture
correspond à la quantité de F1,6-P2 trouvée dans le milieu intracellulaire.
2-7. ETUDE DE L'EFFET DU CAA SUR LE TRANSPORT CELLULAIRE
L'effet de 0, 5, 10, 25 µM CAA sur le transport du glucose dans les cellules MCF-7
est étudié en utilisant le [1-14C]-2-Désoxy-D-glucose (DOG), l'analogue non
métabolisable du glucose.
Des plaques multi puits sont préparées puis incubées de la même façon décrite ci-
dessus. Les dernières 35 minutes d'incubation s'effectuent en présence de DOG. Le
milieu d'incubation, RPMI-1640 avec 5% SVF, est donc remplacé par du milieu
contenant du DOG (0.1 µCi/puits) pendant 35 minutes et toujours en absence ou
présence de 5, 10 ou 25 µM CAA. En fin d'incubation, le milieu est éliminé, les
cellules sont rincées trois fois avec du PBS. Elles sont ensuite solubilisées dans
NaOH 0.1 N. Le nombre total des cellules est déterminé en utilisant des puits
Pour déterminer la radioactivité, les lysats sont récupérés puis ajoutés à 5 ml de
liquide scintillant (
ULTIMA GOLD , PACKARD) dans des fioles à scintillation. Les
comptages sont aussi réalisés sur un aliquote de milieu d'incubation (ce qui
représente le temps zéro). Ceci permet de mesurer la radioactivité spécifique du
DOG ajouté au début d'incubation. Les comptages sont réalisés sur 2 cycles de 5
minutes dans le compteur à scintillation liquide (PACKARD Tri-Carb 2100TR). La
radioactivité obtenue est rapportée au nombre des cellules.
2-8. ETUDE DE L'OXYDATION DU [3,4-14C]-GLUCOSE
Dans les expériences où la production du [14CO2] à partir du [3,4-14C]-glucose a été
mesurée, les cellules sont ensemencées dans des petites boîtes de Pétri ayant une
surface de 9.6 cm2 (surface identique à la surface des puits des plaques à 6 puits).
Les expériences ont été réalisées 48 heures après l'ensemencement de manière à
laisser adhérer et croître les cellules. L'incubation s'effectue dans le milieu RPMI-
1640 contenant du [3,4-14C]-glucose en absence ou en présence d'une concentration
unique de CAA (25 µM). Les conditions expérimentales sont réalisées en double.
Pour réaliser l'incubation, les boîtes de Pétri sont introduites dans des flacons
contenant un puits latéral. Les flacons sont équilibrés avec l'atmosphère de
l'incubateur, fermés d'une manière hermétique puis incubés pendant 24 heures. En
fin d'incubation, et pour arrêter la réaction, l'acide perchlorique (2% v/v,
concentration finale) est ajouté au milieu par une seringue et une aiguille qui traverse
le couvercle du flacon sans l'ouvrir. La soude, qui va capter le CO2 marqué (libéré au
cours de l'incubation) est introduite de la même façon dans le puits latéral.
L'ensemble est laissé à température ambiante pendant une heure. Les flacons sont
ensuite ouverts et le milieu d'incubation est récupéré pour réaliser le dosage de
glucose et de lactate. La soude, qui contient le CO2 marqué, est récupérée pour
mesurer la radioactivité, comme décrit ci-dessus (pages 66 et 69). Le nombre total
des cellules est déterminé en utilisant des puits parallèles.
2-9. ANALYSES STATISTIQUES
Les résultats sont exprimés en moyenne ± erreur standard à la moyenne (SEM).
L'analyse de variance (ANOVA) pour des mesures répétées est appliquée, suivie d'un
test de Fisher pour comparer les résultats des séries expérimentales.
Les analyses statistiques sont effectuées à l'aide d'un logiciel STATVIEW. Des
probabilités inférieures à 0.05 sont considérées comme statistiquement significatives.
2-10. PRODUITS ET MILIEUX
Le Sérum de Veau Fœtal (Fœtal Bovine Serum) est fourni par HyClone, Cramlington,
Le milieu RMPI-1640, l'Amphotericine B, le Pénicilline-Streptomycine la Trypsine-
EDTA sont fournis par Sigma-Aldrich, Germany.
Le PBS (Phosphate-Buffered Saline) est fourni par Invitrogen, USA.
Le [1-14C]-2-Désoxy-D-glucose est fourni par Sigma-Aldrich, Germany.
Le [3,4-14C] glucose est fourni par NEN products, Boston, MA, USA.
1. PREMIERE PARTIE : TOXICITE RENALE DU CAA
1-1. ETUDE DE L'EFFET DOSE DU CAA. DETERMINATION DE LA
CONCENTRATION TOXIQUE DU CAA
L'effet de trois concentrations de CAA est étudié sur le métabolisme du lactate dans
des tranches fines de cortex rénal de rat incubées pendant 24 heures dans le milieu
RPMI-1640 sans D-glucose et sans L-cystine. Les trois concentrations choisies (25,
50 et 75 µM) sont proches de celles mesurées dans le sérum et les urines des
patients traités par l'IFO. Des tranches sont également incubées sans CAA. La
concentration utilisée de lactate (1 mM) est proche de la concentration physiologique.
L'incubation s'effectue dans des rollers dans un incubateur humidifié contenant 40%
d'O2 et 5% de CO2. L'ATP et le glutathion cellulaires, la consommation du lactate, la
néoglucogenèse et l'accumulation du pyruvate sont mesurés.
Les Figures 13 et 14 montrent l'effet du CAA (0, 25, 50 et 75 µM) sur les paramètres
de viabilité cellulaire (l'ATP et le glutathion total).
L'ATP diminue avec toutes les concentrations utilisées de CAA de manière dose
dépendante (54%, 85% et 99% de diminution en présence de 25, 50 et 75 µM CAA,
respectivement par rapport au contrôle sans CAA). A 75 µM CAA, l'ATP est
complètement aboli (0.07 µmol/g protéine). Il en est de même pour le glutathion,
avec un effet plus important dès la plus faible des concentrations (25 µM) où une
diminution d'environ 75% comparé au contrôle (0 µM CAA) est trouvée.
Figure 13 : Effet du CAA sur le contenu d'ATP dans des tranches de cortex rénal incubées
24 h avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=3).
Concentrations de CAA (µM)
Figure 14 : Effet du CAA sur le glutathion total mesuré dans des tranches de cortex rénal
incubées 24 h avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=3).
Concentrations de CAA (µM)
La consommation de lactate, la néoglucogenèse et l'accumulation du pyruvate sont
illustrées dans les Figures 15-17.
Les trois paramètres sont altérés par le CAA mais pas de la même manière. La
quantité de lactate consommé (Figure 15) diminue significativement avec 50 et 75
µM CAA (2629 µmol/g protéines et 2201 µmol/g protéines, respectivement) par
rapport aux tranches témoin incubées sans CAA (3123 µmol/g protéines), tandis que
la quantité de glucose formé (Figure 16) ne diminue significativement qu'avec 75 µM
CAA (61.9 µmol/g protéines) comparé au témoin (131.8 µmol/g protéines).
L'accumulation du pyruvate est le seul paramètre qui soit significativement modifié
avec les trois concentrations utilisées du CAA (Figure 17). Elle augmente de 246.4
µmol/g protéines dans les tranches témoin à 338.8, 507.5 et 583.6 µmol/g protéines
dans les tranches incubées avec 25, 50 et 75 µM CAA, respectivement.
Figure 15 : Effet du CAA sur la consommation de lactate par des tranches de cortex rénal
incubées 24 h avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=3).
µ
é (
m 1500
Concentrations de CAA (µM)
Figure 16 : Effet du CAA sur la néoglucogenèse dans des tranches de cortex rénal incubées
24 h avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=3).
Concentrations de CAA (µM)
Figure 17 : Effet du CAA sur l'accumulation du pyruvate dans des tranches de cortex rénal
incubées 24 h avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=3).
Concentrations de CAA (µM)
Au vu de l'ensemble de ces premiers résultats, la concentration de 75 µM CAA est la
seule qui modifie tous les paramètres étudiés. Elle est donc choisie pour étudier
l'effet du CAA sur les tranches de cortex rénal au cours du temps.
1-2. ETUDE DE L'EFFET DE 75 µM CAA AU COURS DU TEMPS SUR
LE METABOLISME DU LACTATE DANS DES TRANCHES DE
Des tranches fines de cortex rénal de rat sont incubées dans le milieu RPMI-1640
(sans D-glucose et sans L-cystine), contenant 1 mM L-lactate pendant 1, 2, 4, 6, 8, 16
et 24 heures en présence on en absence de 75 µM CAA. L'incubation s'effectue
dans des rollers dans un incubateur humidifié contenant 40% d'O2 et 5% de CO2.
Les paramètres de viabilité des tranches (l'ATP et le glutathion) ainsi que le
métabolisme du lactate (la consommation du lactate, l'accumulation du pyruvate et la
néoglucogenèse) sont étudiés.
Le glutathion baisse rapidement en présence de 75 µM CAA (Figure 18). Dès la
première heure d'incubation nous trouvons 35% de diminution par rapport au témoin
(0 µM CAA) : 4.3 vs. 6.6 µmol/g protéines. L'effet du CAA sur le glutathion s'accroît
au cours du temps. Une diminution du glutathion est également observée au cours
du temps dans les conditions témoin (tranches incubées en absence du CAA). La
concentration du glutathion diminue de 62% après 24 heures d'incubation par rapport
à la concentration mesurée à une heure d'incubation.
Figure 18 : Effet de 75 µM CAA sur le contenu du glutathion dans des tranches de cortex
rénal incubées avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Tem ps d'incubation (heures)
La concentration d'ATP diminue également en présence de 75 µM CAA à partir de 4
heures d'incubation : 7.8 µmol/g protéines vs. 12.1 µmol/g protéines dans le contrôle
sans CAA (soit 36% de diminution) (Figure 19). La chute d'ATP induite par le CAA
devient complète à 16 et à 24 heures d'incubation (0.5 et 0.1 µmol/g protéines,
respectivement). Une diminution de seulement 26% de la concentration d'ATP dans
les tranches incubées en absence de CAA est observée après 24 heures
d'incubation comparativement à la concentration mesurée après une heure
d'incubation, traduisant une bonne viabilité du tissu rénal après 24 heures
Figure 19 : Effet de 75 µM CAA sur le contenu d'ATP dans des tranches de cortex rénal
incubées avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Tem ps d'incubation (heures)
L'effet du CAA est également observé sur la consommation du lactate (Figure 20),
sur l'accumulation du pyruvate (Figure 21) et sur la néoglucogenèse (Figure 22). La
consommation du lactate est significativement inhibée par 75 µM CAA dès 4 heures
d'incubation et d'une manière temps dépendant jusqu'à 24 heures. Parallèlement, on
observe une augmentation de 141% et 152% de l'accumulation du pyruvate en
présence du 75 µM CAA à 16 et à 24 heures d'incubation, respectivement.
Figure 20 : Effet de 75 µM CAA sur la consommation de lactate par des tranches de cortex
rénal incubées avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Tem ps d'incubation (heures)
Figure 21 : Effet de 75 µM CAA sur l'accumulation du pyruvate dans des tranches de cortex
rénal incubées avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Tem ps d'incubation (heures)
La synthèse du glucose par les tranches est inhibée par 75 µM CAA
significativement à partir de 2 heures d'incubation. A 4 heures d'incubation, une
inhibition de 50% de la néoglucogenèse est trouvée : 131.6 µmol/g protéines pour le
témoin contre 65.7 µmol/g protéines en présence de 75 µM CAA. Cette inhibition est
observée tout au long de l'incubation.
Figure 22 : Effet de 75 µM CAA sur la néoglucogenèse dans des tranches de cortex rénal
incubées avec 1 mM lactate. * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Tem ps d'incubation (heures)
Alors que la consommation du lactate et l'accumulation du pyruvate montrent une
augmentation progressive au cours du temps dans les tranches incubées en
absence de CAA, la néoglucogenèse ne montre pas le même profil. La synthèse du
glucose, dans les tranches incubées sans CAA, atteint son niveau maximum à 6
heures d'incubation. Au-delà, le glucose diminue considérablement : 99.4 µmol/g
protéines sont trouvés à 24 heures d'incubation par rapport à 160.9 µmol/g protéines
à 6 heures. Une utilisation du glucose, formé au cours des premières heures
d'incubation, est donc constatée pendant les dernières heures d'incubation (Figure
22). Il est à noter que la présence de 75 µM CAA a inhibé non seulement la
néoglucogenèse, mais également l'utilisation du glucose. La concentration du
glucose trouvée à 24 heures d'incubation n'est pas statistiquement différente de la
concentration maximale du glucose mesurée à 8 heures d'incubation en présence de
L'inhibition de la néoglucogenèse peut être la conséquence de l'inhibition de
certaines activités enzymatiques de cette voie métabolique, ou de la diminution de
l'ATP cellulaire observée en présence du CAA, l'ATP étant nécessaire pour la
synthèse du glucose. Une inhibition de la chaîne respiratoire des mitochondries par
le CAA pourrait être responsable de la diminution d'ATP.
La synthèse du glucose est la voie métabolique la plus sensible à l'action du CAA.
C'est pour cette raison que nous avons décidé d'étudier certaines activités
enzymatiques de cette voie, ainsi que la respiration mitochondriale et l'oxydation du
lactate marqué. Etant donné que l'effet de 75 µM CAA est observé à partir de 4
heures d'incubation sur tous les paramètres étudiés ci-dessus (à l'exception de
l'accumulation du pyruvate), le temps de 4 heures est choisi pour réaliser les études
1-3. EFFET DE 75 µM CAA SUR LES ACTIVITES ENZYMATIQUES DE
LA NEOGLUCOGENESE
Les activités de cinq enzymes de la voie de la néoglucogenèse (Figure 3, page 16)
sont mesurées dans des tranches de cortex rénal de rat incubées 4 heures dans le
milieu RPMI-1640 (sans D-glucose et sans L-cystine) contenant 1 mM L-lactate en
absence ou présence de 75 µM CAA. Les enzymes étudiées sont : la LDH, l'enzyme
qui initie le métabolisme du lactate et sa transformation en pyruvate, la PEPCK, la
F1,6-BPase et la G6-Pase, les trois enzymes qui catalysent les réactions
irréversibles de la néoglucogenèse et la GAPDH, qui possède un groupe thiol dans
son site actif. Le Tableau 2 montre les résultats de ces mesures.
Les activités LDH et PEPCK ne sont pas altérées par le CAA. L'activité GAPDH est
réduite de 53% en présence de 75 µM CAA (de 112.4 à 52.4 UI/g protéines). Le CAA
induit également une inhibition de 26% de l'activité F1,6-BPase (2.3 UI/g protéines
dans le témoin sans CAA contre 1.7 UI/g protéines en présence de 75 µM CAA) et
de 37% de l'activité G6-Pase (19.3 UI/g protéines dans le témoin sans CAA et 12.1
UI/g protéines en présence de 75 µM CAA).
L'inhibition des activités de ces trois enzymes de la néoglucogenèse pourrait donc
expliquer l'inhibition observée de la synthèse du glucose. Il est à noter que l'inhibition
de 53% de la GAPDH est du même ordre de grandeur que l'inhibition de la
néoglucogenèse observée à 4 heures d'incubation (Figure 22).
Pour expliquer l'inhibition de l'activité GAPDH (qui contient un groupe thiol dans son
site actif), le dosage des thiols totaux est effectué pour montrer l'effet du CAA sur ces
composés. Les thiols totaux sont donc mesurés dans des tranches de cortex rénal
incubées 4 heures dans le milieu RPMI-1640 contenant 1 mM L-lactate, en absence
ou en présence de 75 µM CAA. Le Tableau 3 montre que 75 µM CAA induit une
inhibition de 66% du contenu intracellulaire des thiols totaux : 2.6 µmol/g protéines
en présence du CAA vs. 7.7 µmol/g protéines en absence du CAA.
Tableau 3 : Effet de 75 µM CAA sur le contenu cellulaire des thiols totaux dans des tranches
de cortex rénal de rat incubées 4 h avec 1 mM lactate. Valeurs présentées sous forme de
moyenne ± SEM. * : P< 0.05 vs. contrôle (0 µM CAA), (n=4).
Thiols totaux
(µmol/g protéines)
1-4. EFFET DE 75 µM CAA SUR LA RESPIRATION
MITOCHONDRIALE ET L'OXYDATION DU [U-14C]-LACTATE
La respiration mitochondriale est étudiée dans des mitochondries isolées des
tranches de cortex rénal préalablement incubées pendant 4 heures dans le milieu
RPMI-1640 (sans D-glucose et sans L-cystine) contenant 1 mM L-lactate en présence
ou en absence de 75 µM CAA. Les résultats sont montrés dans le Tableau 4. La
consommation d'oxygène est mesurée en présence de plusieurs substrats. Les
mélanges (malate + pyruvate), et (malate + glutamate) sont des donneurs d'électrons
pour former le NADH qui sera re-oxydé en NAD+ au niveau du complexe I de la
chaîne respiratoire. Le succinate donne les électrons nécessaires pour la formation
du FADH2, re-oxydé en FAD au niveau du complexe II.
Au niveau du complexe II, la consommation d'oxygène n'est pas inhibée en présence
de 75 µM CAA, ce qui implique également l'intégrité des complexes III et IV de la
chaîne respiratoire. Au contraire, la consommation d'oxygène au niveau du complexe
I est inhibée en présence de 75 µM CAA de 22% dans le cas où le mélange (malate
+ pyruvate) est le donneur d'électrons et de 23% dans le cas où le mélange (malate
+ glutamate) est utilisé.
Tableau 4 : Effet de 75 µM CAA sur la consommation d'oxygène au niveau des complexes I
et II de la chaîne respiratoire des mitochondries isolées des tranches de cortex rénal de rat
préalablement incubées pendant 4 h avec 1 mM lactate. Valeurs présentées sous forme de
moyenne ± SEM. * : P< 0.05 vs. contrôle (0 µM CAA), (n=7).
Consommation d'oxygène
(nano-atom O/mg protéines/minute)
Complexe I
Complexe II
Malate + Pyruvate
Malate + Glutamate
L'oxydation du [U-14C]-lactate en [14CO2] via la PDH et le cycle des acides
tricarboxylique est mesurée dans des tranches de cortex rénal incubées pendant 4
heures dans le milieu RPMI-1640 (sans D-glucose et sans L-cystine) contenant 1 mM
de [U-14C]-lactate en présence ou en absence de 75 µM CAA. Les résultats sont
présentés dans le Tableau 5.
Tableau 5 : Effet de 75 µM CAA sur la formation de [14CO2] par des tranches de cortex rénal
de rat incubées 4 h avec 1 mM de [U-14C]-lactate. Valeurs présentées sous forme de
moyenne ± SEM. * : P< 0.05 vs. contrôle (0 µM CAA), (n=6).
Formation de [14CO2]
(µmol/g protéines)
Le CAA induit une inhibition de 19% de la formation de [14CO2] à partir de [U-14C]-
lactate. Cette inhibition est du même ordre de grandeur que l'inhibition observée du
complexe I de la chaîne respiratoire.
Les résultats de cette première partie montrent un effet toxique du CAA sur les
tranches de cortex rénal. L'inhibition des activités enzymatiques de la
néoglucogenèse (notamment la GAPDH), et de la respiration mitochondriale (au
niveau du complexe I de la chaîne respiratoire) semblent être des mécanismes
responsables de cet effet. L'inhibition par le CAA de la GAPDH, qui est à la fois une
enzyme de la néoglucogenèse et de la glycolyse, pourrait provoquer une inhibition
de ces deux voies métaboliques. Etant donné que les cellules cancéreuses utilisent
de grosses quantités de glucose par la voie glycolytique, nous nous sommes posés
la question de savoir si le CAA pouvait, en inhibant cette voie, altérer la croissance
des cellules cancéreuses. C'est l'objet de la deuxième partie dont les résultats sont
présentés ci-après.
2. DEUXIEME PARTIE : EFFET DU CAA SUR LES CELLULES
DE CANCER DU SEIN (MCF-7)
2-1. EFFET DU CAA SUR LA PROLIFERATION CELLULAIRE
L'effet du CAA sur la prolifération des cellules MCF-7 incubées pendant 24 heures
dans le milieu RPMI-1640 contenant 5% SVF, en absence ou en présence des
différentes concentrations de CAA (5, 10, 25 et 50 µM), est étudié en déterminant le
nombre total et la viabilité des cellules. Les concentrations utilisées de CAA sont
proches de celles trouvées chez des patients traités par l'IFO [33, 96, 109-111]. Les
résultats sont montrés dans le Tableau 6.
Tableau 6 : Effet du CAA sur le nombre total et la viabilité des cellules MCF-7 incubées 24 h.
ND : non déterminé. * : P< 0.05 vs. contrôle (0 µM CAA), (n=10).
Concentration du CAA Nombre total de cellules
Cellules mortes
(% du nombre total des cellules)
Le nombre des cellules MCF-7 augmente de 1.31 ± 0.06 x106 cellules/puits dans le
contrôle à 1.88 x106 cellules/puits après 24 heures d'incubation en absence de CAA.
Dix µM CAA inhibe significativement la croissance cellulaire (1.54 x106 cellules/puits).
L'effet inhibiteur du CAA sur la croissance des cellules est maximal avec 25 µM CAA
(1.26 x106 cellules/puits). Il faut mentionner qu'avec une concentration plus forte de
CAA (50 µM), les cellules étaient presque complètement détruites (0.03 x106
cellules/puits). La viabilité des cellules, déterminée en utilisant l'iodure de propidium,
n'est pas modifiée en présence de 5 et 10 µM CAA. Elle diminue quand la
concentration de 25 µM CAA est appliquée (environ 20 % des cellules mortes).
Les photos des cellules, prises à l'aide d'un microscope et d'un logiciel, montrent
l'effet du CAA sur la prolifération cellulaire (Figure 23). La morphologie des cellules
indique, malgré la diminution du nombre total, qu'elles sont vivantes en présence de
5, 10 et 25 µM CAA. En présence de 50 µM CAA, le peu de cellules trouvées
deviennent rondes et flottent dans le milieu. Les concentrations "cliniques" de CAA
inhibent donc, la prolifération des cellules MCF-7.
Figure 23 : Photos des cellules MCF-7 prises après 24 h d'incubation. Grossissement x10
A : sans CAA, B : avec 5 µM CAA, C : avec 10 µM CAA, D : avec 25 µM CAA et E : avec 50
2-2. EFFET DU CAA SUR LE METABOLISME DU GLUCOSE
La glycolyse est la voie métabolique principale des cellules cancéreuses. Nous
avons étudié cette voie dans des cellules MCF-7 incubées pendant 24 heures dans
le milieu RPMI-1640 en absence ou en présence de 3 concentrations de CAA (5, 10
et 25 µM). L'utilisation du glucose et la production du lactate sont mesurées. Etant
donné que les cellules étaient quasiment complètement détruites en présence de 50
µM CAA, ces deux paramètres n'ont pas été étudiés en présence de cette
La Figure 24 montre que l'utilisation du glucose diminue significativement à partir de
10 µM CAA (3.6 µmol/106 cellules vs. 4.6 µmol/106 cellules dans le contrôle sans
CAA). Elle est inhibée de 45% par 25 µM CAA (2.6 µmol/106 cellules).
Figure 24 : Effet du CAA sur l'utilisation du glucose dans les cellules MCF-7 incubées 24 h
dans le milieu RPMI-1640 (5% SVF). * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Concentrations du CAA (µM)
La production du lactate, le produit principal du métabolisme du glucose dans les
cellules cancéreuses, diminue également en présence de CAA (Figure 25). Cette
diminution n'est significative qu'avec 25 µM CAA (3.9 µmol/106 cellules vs. 6.3
µmol/106 cellules dans le contrôle sans CAA, soit 38% d'inhibition). Cette inhibition
de la production du lactate est du même ordre de grandeur que l'inhibition de
l'utilisation de glucose. Il est à noter que le ratio "lactate produit/glucose utilisé" n'est
que peu modifié en présence de 25 µM CAA par rapport au contrôle sans CAA (1.52
Figure 25 : Effet du CAA sur la production du lactate dans les cellules MCF-7 incubées 24 h
dans le milieu RPMI-1640 (5% SVF). * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Concentrationc du CAA (µM)
Etant donné l'importance de cette voie métabolique dans les cellules cancéreuses,
l'inhibition de la glycolyse (l'utilisation du glucose) par le CAA pourrait donc expliquer
l'inhibition de la prolifération cellulaire.
2-3. EFFET DU CAA SUR L'ENERGIE CELLULAIRE
La plupart des données de la littérature considèrent la glycolyse comme la source
principale de la production d'énergie dans les cellules cancéreuses. L'ATP cellulaire
est donc mesuré dans des cellules MCF-7 incubées 24 heures dans le milieu RPMI-
1640 en absence ou en présence de 5, 10 et 25 µM CAA (Figure 26). Le niveau
d'ATP cellulaire ne change pas en présence de 5 et 10 µM CAA par rapport au
contrôle. D'une manière surprenante, l'ATP augmente dans les cellules incubées
avec 25 µM CAA, une augmentation de 54% par rapport au contrôle sans CAA (13.6
vs. 8.8 nmol/106 cellules, respectivement).
Figure 26 : Effet du CAA sur l'ATP des cellules MCF-7 incubées 24 h dans le milieu RPMI-
1640 (5% SVF). * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Concentrations du CAA (µM)
Il est donc évident que l'inhibition de la prolifération cellulaire, induite par le CAA, n'a
pas pour cause la chute d'ATP cellulaire, ce dernier n'étant pas diminué en présence
de 25 µM CAA. Par ailleurs, la glycolyse n'est pas la source unique d'énergie
cellulaire puisque la concentration d'ATP ne diminue pas malgré une inhibition de
45% de la glycolyse en présence de 25 µM CAA.
2-4. EFFET DU CAA SUR LA PRODUCTION DE [14CO2] A PARTIR DE
[3,4-14C]-GLUCOSE
Etant donné que l'inhibition de la glycolyse n'a pas engendré une diminution de l'ATP,
et pour vérifier une source oxydative éventuelle de l'énergie cellulaire, nous avons
mesuré la quantité de glucose complètement oxydée dans les cellules MCF-7
incubées dans les mêmes conditions mentionnées ci-dessus. La quantité de glucose
complètement oxydée est calculée en mesurant la conversion de [3,4-14C]-glucose
en [14CO2] via la réaction catalysée par la pyruvate déshydrogénase. L'effet de 25
µM CAA sur cette quantité est étudié. Comme le montre le Tableau 7, environ 6% du
glucose utilisé est complètement oxydé sans que ce pourcentage ne soit modifié par
la présence de 25 µM CAA.
Tableau 7 : Mesure de la conversion de [3,4-14C]-glucose en 14CO2 par les cellules MCF-7
incubées pendant 24 h dans le milieu RPMI-1640 (5% SVF) en absence ou en présence de
25 µM CAA. * : P< 0.05 vs. contrôle (0 µM CAA), (n=4).
Concentration du CAA (µM)
Glucose utilisé (µmol/106 cellules ± SEM)
Glucose complètement oxydé
(µmol/106 cellules ± SEM)
Glucose oxydé / glucose utilisé (%)
2-5. EFFET DU CAA SUR LES ACTIVITES ENZYMATIQUES DE LA
La glycolyse est inhibée par le CAA (Figure 24). Les activités de certaines enzymes
de cette voie métabolique sont mesurées dans les cellules MCF-7 incubées 24
heures dans le milieu RPMI-1640 en présence des différentes concentrations de
CAA (5, 10 et 25 µM). Les trois enzymes clés de la glycolyse sont l'HK, la PFK, et la
PK. Elles catalysent les trois réactions irréversibles de la glycolyse. Leurs activités
ainsi que celles de la GAPDH et la G6PDH sont étudiées (voir Figure 4, page 19).
L'activité HK, qui initie la voie glycolytique, diminue en présence de 25 µM CAA (8.5
mUI/106 cellules vs. 12.0 mUI/106 cellules dans le contrôle sans CAA) (Figure 27-A).
L'activité PK diminue également sous l'effet de 25 µM CAA (307.3 mUI/106 cellules
vs. 412.5 mUI/106 cellules dans le contrôle sans CAA) (Figure 27-C). Le CAA ne
montre pas d'effet sur l'activité PFK (Figure 27-B). L'activité HK est la plus faible des
trois activités clés dans le contrôle : 12.0 mUI/106 cellules vs. 43.0 mUI/106 cellules
pour la PFK et 412.5 mUI/106 cellules pour la PK.
Comme nous l'avons précédemment mentionné, la GAPDH est à la fois une enzyme
de la glycolyse et de la néoglucogenèse. Sous l'effet de 75 µM CAA sur des tranches
de cortex rénal (incubées pendant 4 heures), l'activité GAPDH était inhibée de 53%.
Dans les cellules MCF-7, l'activité GAPDH est inhibée en présence de 10 et 25 µM
CAA (18% et 62% d'inhibition par rapport au contrôle sans CAA, respectivement)
La G6PDH catalyse la première étape de la voie des pentoses phosphate à partir du
G6-P. L'effet du CAA sur l'activité G6PDH est montré dans la Figure 29. Cette activité
n'est pas modifiée en présence de 25 µM CAA. Au contraire, elle augmente sous
l'effet de 5 et 10 µM CAA (186.9 et 169.9 mUI/106 cellules, respectivement) comparé
au contrôle sans CAA (136.5 mUI/106 cellules).
Figure 27 : Effet du CAA sur les activités (A) : hexokinase (HK), (B) : phosphofructokinase
(PFK) et (C) : pyruvate kinase (PK) dans les cellules MCF-7 incubées 24 h dans le milieu
RPMI-1640 (5% SVF). * : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
Concentrations du CAA (µM)
les)
lu 30
cel
Concentrations du CAA (µM)
lu 300
cel
Concentrations du CAA (µM)
Figure 28 : Effet du CAA sur l'activité glyceraldéhyde 3-phosphate déshydrogénase
(GAPDH) dans les cellules MCF-7 incubées 24 h dans le milieu RPMI-1640 (5% SVF).
* : P< 0.05 vs. contrôle (0 µM CAA), (n=5).
DH ( 100
AP
G
Concentrations du (µM)
Figure 29 : Effet du CAA sur l'activité glucose 6-phosphate déshydrogénase (G6PDH) dans
les cellules MCF-7 incubées 24 h dans le milieu RPMI-1640 (5% SVF). * : P< 0.05 vs.
contrôle (0 µM CAA), (n=5).
Concentrations du CAA (µM)
2-6. EFFET DU CAA SUR LE TRANSPORT CELLULAIRE DU
Dans les cellules cancéreuses, le transport du glucose est une étape essentielle afin
d'initier son métabolisme. L'inhibition du métabolisme du glucose dans les cellules
MCF-7 peut résulter, en plus de l'inhibition de certaines activités enzymatiques de la
glycolyse, d'un effet inhibiteur du CAA sur le transport du glucose.
Le transport du [1-14C]-2-Désoxy-D-glucose (DOG), un analogue non métabolisable
du glucose, est étudié dans des cellules MCF-7 préalablement incubées pendant 24
heures dans le milieu RMPI-1640 en présence des différentes concentrations de
CAA (5, 10 et 25 µM). Le DOG est ajouté au milieu pendant 35 minutes, son
incorporation à l'intérieur des cellules est ensuite mesurée (Figure 30).
Figure 30 : Effet du CAA sur le transport du [1-14C]-2-Désoxy-D-glucose (DOG) étudié
pendant 35 minutes dans des cellules MCF-7 pré incubées 24 h dans le milieu RPMI-1640
(5% SVF). Valeurs présentés sous forme de coups par minute (CPM) et par nombre de
cellules. * : P< 0.05 vs. contrôle (0 µM CAA), (n=4).
cel60/1 6000
Concentrations du CAA (µM)
Le transport du DOG est inhibé pour toutes les concentrations étudiées de CAA de
manière dose dépendante : 17%, 32% et 33% d'inhibition en présence de 5, 10 et 25
µM CAA, respectivement, par rapport au contrôle sans CAA.
L'inhibition de la glycolyse peut donc être expliquée, en partie, par l'inhibition du
transport du glucose, qui s'additionne à l'inhibition de l'activité HK, l'enzyme limitante
de la voie glycolytique.
2-7. EFFET DU CAA SUR LES CELLULES MCF-7 EN HYPOXIE (5%
Les résultats présentés ci-dessus montrent que le CAA inhibe la glycolyse et la
prolifération des cellules MCF-7 incubées en normoxie. Cette inhibition n'est pas
accompagnée d'une diminution de l'énergie cellulaire (le niveau d'ATP cellulaire ne
diminue pas en présence de CAA). L'ATP provient donc d'autres sources que la
glycolyse, probablement la voie oxydative.
L'hypoxie est une caractéristique importante de l'environnement tumoral, et surtout
les tumeurs solides. Dans l'étude suivante, les cellules MCF-7 sont incubées en
hypoxie afin de vérifier si celle-ci renforce l'effet du CAA sur les cellules en inhibant
l'ATP d'origine oxydative.
2-7-1. Effet du CAA sur la prolifération cellulaire
La modification du nombre total des cellules MCF-7 induite par les différentes
concentrations du CAA en hypoxie montre le même profil que celui obtenu en
normoxie (Tableau 8). Le nombre des cellules augmente de 1.37 ± 0.02 x 106
cellules à 1.82 x 106 cellules après 24 heures d'incubation en absence de CAA. La
croissance cellulaire est inhibée en présence de 10 et 25 µM CAA comparé au
contrôle sans CAA. Les valeurs obtenues en hypoxie sont comparables à celles
obtenues en normoxie pour toutes les concentrations utilisées (Tableau 6, page 91).
Tableau 8 : Effet du CAA sur le nombre total des cellules MCF-7 incubées 24 h en hypoxie
(5% O2). ND : non déterminé. * : P< 0.05 vs. contrôle (0 µM CAA), (n=4).
Nombre total de cellules
Cellules mortes
du CAA (µM)
(% du nombre total des cellules)
2-7-2. Effet du CAA sur le métabolisme du glucose et l'énergie cellulaire
L'effet du CAA (5, 10 et 25 µM) sur le métabolisme du glucose (consommation du
glucose et production de lactate) et sur l'ATP cellulaire est étudié dans les cellules
MCF-7 incubées pendant 24 heures en hypoxie (5% O2) dans le milieu RPMI-1640.
Les résultats sont montrés dans le Tableau 9.
Tableau 9 : Effet du CAA sur le métabolisme du glucose (consommation du glucose et
production du lactate) et sur l'ATP dans les cellules MCF-7 incubées 24 h en hypoxie (5%
O2). Valeurs présentées sous forme de moyenne ± SEM. * : P< 0.05 vs. contrôle (0 CAA),
Glucose consommé Lactate produit
ATP cellulaire
du CAA (µM)
(µmol/106 cellules)
(µmol/106 cellules)
(nmol/106 cellules)
Le CAA, en hypoxie, induit le même profil d'inhibition que celui observé en normoxie
sur la consommation du glucose (inhibée de 20% et 53% par 10 et 25 µM CAA,
respectivement par rapport au contrôle sans CAA) et sur la production du lactate
(inhibée de 48% en présence de 25 µM CAA, comparé au contrôle). Il est à noter
qu'en hypoxie, la consommation du glucose ainsi que la production du lactate dans
les conditions témoin sans CAA sont environ deux fois plus élevées qu'en normoxie
(8.3 vs 4.6 µmol/106 cellules et 13.5 vs 6.3 µmol/106 cellules, respectivement). Le
ratio lactate produit / glucose utilisé n'est guère plus élevé en présence de 25 µM
CAA qu'en absence du CAA (1.78 vs. 1.64). Ce même profil était déjà observé en
normoxie (1.52 vs. 1.36).
Il en est du même pour l'ATP cellulaire. En hypoxie, l'effet du CAA sur l'ATP montre
le même profil qu'en normoxie (Tableau 9). Le contenu d'ATP ne change pas en
présence de 5 et 10 µM CAA. Il augmente de 23% en présence de 25 µM CAA (10.8
nmol/106 cellules) comparé au contrôle sans CAA (8.8 nmol/106 cellules). Cette
augmentation est moins importante que celle observée en présence de 25 µM CAA
en normoxie (54%).
2-7-3. Effet du CAA sur les activités enzymatiques de la glycolyse
Le même profil d'effet du CAA sur les activités enzymatiques étudiées en normoxie
est retrouvé en hypoxie (Tableau 10). Comparées au contrôle sans CAA, les activités
HK et PK sont inhibées par 25 µM CAA tandis que l'activité PFK ne change pas.
L'activité GAPDH est également inhibée par 25 µM CAA. L'activité G6PDH augmente
en présence de 5 et 10 µM CAA.
En comparant les valeurs des activités enzymatiques dans le contrôle sans CAA, il
est évident que l'hypoxie induit l'expression de toutes les enzymes mesurées, à
l'exception de la G6PDH, probablement sous l'effet d'HIF-1 (Figures 27-29, pages 99-
L'hypoxie (5% O2) ne renforce donc pas l'effet du CAA sur les cellules MCF-7. Nous
avons obtenu les mêmes profils en hypoxie qu'en normoxie pour tous les paramètres
étudiés (la prolifération des cellules, le métabolisme du glucose et les activités
enzymatiques). Même l'ATP n'a pas diminué en hypoxie. Les cellules incubées en
absence de CAA ont un taux élevé d'ATP tout à fait comparable à celui obtenu en
normoxie. La présence de 5 et 10 µM CAA n'a pas altéré l'ATP en hypoxie. La
concentration de 25 µM CAA a, au contraire, augmenté le taux d'ATP, une
augmentation déjà observée en normoxie.
En conclusion, l'hypoxie appliquée dans nos expériences, induit une activation du
métabolisme du glucose et une surexpression des enzymes de la glycolyse que nous
avons étudiées, mais elle n'était peut-être pas suffisamment sévère pour inhiber la
phosphorylation oxydative dans les cellules MCF-7.
1. VALIDATION DU MODELE EXPERIMENTAL
1-1. LES TRANCHES DE CORTEX RENAL
Le modèle de tranches de tissus est ancien (utilisé depuis plus de 60 ans). Il a été
largement appliqué dans des études biochimiques, pharmacologiques et
toxicologiques [232]. Les autres modèles d'études in vitro (les cellules isolées et les
cellules en culture) l'ont remplacé à cause de sa mauvaise reproductibilité et la
limitation du nombre de tranches pouvant être obtenues.
Depuis l'élaboration des appareils automatisés dans les années 1980, comme le
microtome de Krumdieck [233] et le microtome de Brendel-Vitron [232], le modèle de
tranche tissulaire jouit d'un regain d'intérêt, et de nombreux horizons ont été ouverts
pour l'utilisation de ces tranches [234-237]. Ces microtomes permettent d'obtenir des
tranches fines et reproductibles ayant une épaisseur et un diamètre calibrés à 200-
250 µm et 5 mm, respectivement. Grâce à ce système, les tranches sont obtenues
assez rapidement. Le recours à un système d'incubation en roller a permis des
incubations sur des périodes longues (plusieurs jours) ce qui représente un intérêt
majeur pour les études de toxicologie. Ce modèle est appliqué essentiellement sur le
foie [235]. Il est également utilisé pour d'autres organes comme le rein, le poumon, le
cœur, l'intestin et l'ovaire [236, 238].
L'incubation des tranches déposées sur la grille des rollers, dans une fiole contenant
le milieu de culture, et dans une atmosphère riche en O2, nous a permis de réaliser
des incubations jusqu'à 24 heures. La survie du tissu est maintenue pendant cette
période avec conservation de la structure et de la différenciation tissulaire. Le
système de rollers permet aux tranches d'être alternativement en contact avec le
milieu d'incubation et avec le mélange gazeux.
Les avantages des tranches tissulaires par rapport aux cellules en culture ou des
tubules isolés sont multiples. Le tissu est obtenu sans emploi d'enzymes
protéolytiques. Les tranches peuvent être utilisées ou conservées pendant des
périodes assez longues (plusieurs jours) contrairement aux cellules isolées dont le
temps de survie est de quelques heures tout au plus. Par rapport aux cellules en
culture, il n'y a pas de phénomène de dédifférenciation qui apparaît lorsque les
cellules fraîchement isolées commencent à croître après 2 ou 3 jours de culture.
Enfin, l'architecture cellulaire, c'est-à-dire les interactions cellules-cellules et cellules-
matrice sont préservées dans le modèle de tranches. Un point faible du modèle de
tranches par rapport aux cellules isolées est la moins grande intensité du
métabolisme due en grande partie à la moins bonne accessibilité des substrats
exogènes vers les cellules.
Dans notre étude, une concentration minimale d'ATP de 6.2 µmol/g protéines
mesurée après 24 heures d'incubation en rollers est tout à fait satisfaisante. Elle
montre que les tranches de cortex rénal coupées avec précision sont viables d'un
point de vue métabolique, malgré une chute du glutathion à 24 heures d'incubation.
Cette concentration est proche de celle de 6.9 µmol/g protéines trouvée dans une
autre étude réalisée dans notre laboratoire qui avait également montré une faible
libération de LDH dans le milieu extracellulaire (1.5%) après 24 heures d'incubation
[239]. Cette concentration est également proche de celle mesurée, dans des tubules
isolés de rein humain, par Dubourg et al. (8 µmol/g protéines) [115].
L'utilisation de ce modèle dans la première partie de notre étude est donc justifiable.
Il remplace le modèle de tubules isolés sur lequel les études de la toxicité du CAA
ont été précédemment réalisées [113, 115]. Il est choisi pour étudier l'effet
néphrotoxique du CAA pendant des périodes jusqu'à 24 heures. L'augmentation du
temps d'exposition du tissu rénal au CAA (24 heures pour les tranches contre 1
heure pour les tubules) nous a permis d'utiliser de faibles concentrations du toxique
pour obtenir une néphrotoxicité. Ces concentrations sont très proches de celles
mesurées dans le sang et les urines des patients traités par l'IFO [33, 96, 109-111],
soit 0-75 µM. Les tranches métabolisent du lactate, un substrat physiologique du rein,
présent en concentration de 1 mM, proche de la concentration physiologique du
1-2. LES CELLULES MCF-7 DE CANCER DU SEIN
Dans la deuxième partie de notre étude, nous avons recherché un effet toxique
éventuel du CAA sur les cellules MCF-7 de cancer du sein. Nous avons voulu savoir
si le CAA pourrait contribuer à l'effet thérapeutique de l'IFO.
L'utilisation des cellules MCF-7 dans la recherche sur le cancer est largement
répandue. C'est la lignée de cellules tumorales la plus utilisée dans les laboratoires
de recherche sur le cancer du sein. Elle est mentionnée dans plus d'un tiers des
articles rapportant des travaux sur des lignées tumorales mammaires (environ 11000
références sur PubMed). Cette lignée est souvent utilisée dans les recherches de
nouveaux médicaments anticancéreux, les études de la toxicité de certains agents
chimiothérapeutiques et les applications d'hormonothérapie pour le cancer du sein
[182, 186, 208, 209, 240].
La manipulation et l'entretien de cette lignée s'effectuent, sans trop de difficulté, dans
un milieu RPMI-1640 contenant du sérum de veau fœtal. Un antibiotique et un
antifongique sont ajoutés au milieu. Aucune contamination n'est survenue durant
toute l'étude. Tout au long de nos expériences, nous avons constaté une
reproductibilité excellente des conditions expérimentales. L'exposition des cellules au
CAA a toujours été réalisée au même degré de confluence. Durant les 24 heures
d'expérience, la croissance des cellules témoin était importante (45% environ), reflet
d'un bon fonctionnement cellulaire. L'utilisation de ce modèle est donc justifiable.
1-3. LES DOSAGES
Les différents dosages, enzymatiques et radioactifs, sont parfaitement spécifiques.
Les méthodes enzymatiques ont été validées par la plupart des livres de biochimie.
2. PREMIERE PARTIE : TOXICITE RENALE DU CAA
2-1. ETUDE DE L'EFFET DOSE DU CAA. DETERMINATION DE LA
CONCENTRATION TOXIQUE DU CAA
Dans cette partie, nous avons étudié l'effet néphrotoxique du CAA sur le modèle de
tranches de cortex rénal. Cet effet a été démontré par plusieurs auteurs sur d'autres
modèles expérimentaux [76, 113, 114]. Dans un premier temps, nous avons utilisé
trois concentrations de CAA (25, 50 et 75 µM). Nous avons incubé les tranches en
présence ou en absence de ces concentrations pendant 24 heures. Les tranches
métabolisaient 1 mM lactate comme substrat physiologique. L'incubation a eu lieu
sur des rollers dans un incubateur multigaz (40% d'O2 et 5% de CO2) saturé
d'humidité. Le milieu d'incubation est du RPMI-1640 sans D-glucose et L-cystine. La
cystine est un thiol connu pour sa capacité à former des liaisons chimiques avec le
CAA l'empêchant d'exercer son effet [116], ce qui justifiait l'utilisation d'un tel milieu
afin de ne pas masquer l'effet du toxique.
Les paramètres de viabilité cellulaire (l'ATP et le glutathion) ont été mesurés en fin
d'incubation. Nos résultats montrent l'effet toxique du CAA sur ces paramètres de
manière dose dépendante. Dès la plus faible des concentrations (25 µM), la
concentration d'ATP diminue de 54% comparée au contrôle. En présence de 75 µM
CAA, on ne trouve quasiment plus d'ATP dans les tranches (0.07 µmol/g protéines).
Il en est de même pour le glutathion qui s'avère plus sensible que l'ATP. Il diminue
de 73% en présence de 25 µM CAA. Ceci confirme les résultats obtenus par d'autres
auteurs sur la sensibilité du glutathion au CAA [73, 80, 121].
La déplétion du glutathion, le composé thiol principal dans la cellule, pourrait être à
l'origine da la néphrotoxicité du CAA, celui-ci étant capable de se lier directement au
glutathion pour former le 2-(s-chloroéthyl)-GSH [241], conduisant à la diminution des
concentrations de ce composé. Lind et al. [116] avaient déjà montré que le CAA est
un composé très réactif qui peut, à concentration équimolaire, se lier aux composés
thiols de manière chimique non enzymatique. Dubourg et al. ont confirmé ces
observations [113].
Le glutathion joue un rôle important dans de nombreuses fonctions biologiques
comme la détoxication, la protection contre le stress oxydatif (rôle antioxydant) et le
maintien du potentiel redox cellulaire [242, 243]. Il a été déjà montré que l'atteinte
rénale induite par le CAA est accompagnée d'une dégradation du glutathion, d'une
augmentation de la péroxydation des lipides et d'une diminution d'ATP cellulaire [73,
77, 80]. Cette atteinte peut se manifester chez certains patients par un syndrome de
L'ajout du mesna et d'amifostine, deux composés qui contiennent des groupes SH
connus pour avoir une capacité protectrice des thiols, avait été proposé par plusieurs
auteurs pour protéger des effets toxiques du CAA. Le mesna n'avait pas prévenu la
toxicité rénale d'IFO [51, 67, 68]. Hartmann et al. [70] ont montré que les patients
traités par l'amifostine développent une atteinte rénale moins sévère que les autres
patients. Cependant, d'autres études ont montré l'absence de cet effet protecteur
d'amifostine contre la néphrotoxicité de l'IFO in vivo [72, 244] et in vitro [69].
Pour confirmer cet effet toxique, le métabolisme du lactate 1 mM dans les tranches
est étudié en absence ou en présence des trois concentrations mentionnées du CAA.
Nous avons mesuré la consommation du lactate, la synthèse du glucose et
l'accumulation du pyruvate.
Le métabolisme rénal du lactate est inhibé par le CAA de manière dose dépendante.
Nous observons une diminution du lactate consommé en présence de 50 et 75 µM
CAA (de 16% et 30%, respectivement) et une inhibition de 53% de la synthèse du
glucose avec 75 µM CAA par rapport au témoin. Le pyruvate est le seul paramètre
du métabolisme du lactate qui soit modifié significativement avec toutes les
concentrations utilisées. Sous l'effet de 75 µM CAA, la quantité accumulée de
pyruvate est plus de deux fois supérieure à celle du témoin.
L'accumulation du pyruvate peut signifier une inhibition de l'activité des deux
enzymes impliquées dans son métabolisme : la PC qui initie la néoglucogenèse à
partir du pyruvate et la PDH qui catalyse la première étape de l'oxydation du
pyruvate. L'inhibition de l'oxydation explique la diminution observée ci-dessus de la
concentration d'ATP. Les activités PC et PDH n'ont pas été mesurées dans notre
étude mais il a été déjà montré par Dubourg et al. [115], dans l'étude réalisée dans
notre laboratoire sur des tubules isolés, que le CAA induit une déplétion de la
concentration intracellulaire de CoA et d'acétyl CoA. Le CoA est le substrat de la
réaction catalysée par la PDH, et l'acétyl CoA est un activateur de la PC [245].
En outre, l'accumulation observée du pyruvate, favorise le déplacement de l'équilibre
de la réaction catalysée par la LDH vers la formation de lactate et, par conséquent,
diminue l'utilisation du lactate afin de maintenir le rapport lactate/pyruvate constant.
Ceci explique en partie l'inhibition da la consommation du lactate en présence du
CAA. Comme nous en discuterons plus tard, cette inhibition de la consommation de
lactate est également due à l'inhibition de plusieurs enzymes de la néoglucogenèse
ainsi qu'à l'inhibition de la chaîne respiratoire mitochondriale.
Contrairement à l'étude déjà réalisée dans notre laboratoire, où les effets toxiques du
CAA étaient observés sur les tubules rénaux isolés à forte concentration de toxique,
soit 500 µM, [113], nous avons montré cet effet toxique en utilisant des
concentrations beaucoup plus faibles de CAA, proches de celles mesurées dans le
sang et les urines des patients traités par l'IFO, qui variaient entre 5 et 200 µM selon
l'étude [33, 96, 109-111]. L'explication de cette différence provient de la différence de
modèle utilisé. Les tubules rénaux sont viables pendant 1 ou 2 heures seulement, et
ils ne peuvent pas être incubés pour des périodes longues. Il fallait donc appliquer le
CAA à des concentrations élevées pour induire un effet toxique. L'utilisation des
tranches dans notre étude nous a permis de surmonter le problème de
concentrations et de montrer l'effet toxique de concentrations "cliniques" du CAA sur
des périodes assez longues.
Nos résultats confirment que le CAA est néphrotoxique, ce qui a été déjà montré par
d'autres auteurs sur un modèle de rein isolé perfusé [78, 80] ou sur des cellules
rénales en culture [112]. Au vu de l'ensemble de ces premiers résultats, la
concentration de 75 µM CAA est la seule qui engendre une modification de tous les
paramètres étudiés. Elle est donc choisie pour étudier l'effet du CAA sur les tranches
au cours du temps.
2-2. ETUDE DE L'EFFET DE 75 µM CAA AU COURS DU TEMPS SUR
LE METABOLISME DU LACTATE DANS DES TRANCHES DE
Nous avons étudié l'effet de 75 µM CAA au cours du temps sur les paramètres de
viabilité cellulaire et sur le métabolisme de 1 mM lactate dans des tranches de cortex
rénal incubées pendant 1, 2, 4, 6, 8, 16 et 24 heures. Au bout d'une heure
d'incubation, aucune modification des paramètres étudiés n'a été constatée, à
l'exception d'une diminution de 35% du glutathion. Cette inhibition confirme la
sensibilité déjà observée de ce paramètre au CAA. L'effet du CAA sur le glutathion
est plus important à partir de 2 heures d'incubation et il s'accroît au cours du temps
jusqu'à 24 heures. Il est à noter que la dégradation du glutathion apparaît au cours
du temps même en absence de CAA au delà de 16 heures d'incubation. Ainsi on ne
trouve, à 24 heures, que 38% de la valeur du glutathion trouvée au début
d'incubation (à une heure). Cela montre que l'incubation, en elle-même, induit des
dégâts sur les tranches. Dans ce contexte, il est utile de rappeler que nos
expériences ont été réalisées dans un milieu sans L-cystine afin de prévenir la
fixation non spécifique du CAA par ce composé thiol. La diminution du glutathion
observée en absence de CAA pourrait être attribuée à l'absence de sa régénération
due au manque des précurseurs des composés thiols. Malgré tout, l'effet du CAA sur
le contenu du glutathion dans les tranches est observé même à 16 et à 24 heures.
L'ATP est inhibé par 75 µM CAA, de façon significative à partir de 4 heures
d'incubation. L'inhibition est quasiment totale à 16 et à 24 heures. Cela confirme
l'effet toxique du CAA sur les tranches déjà observé sur le glutathion. Nous avons
noté une augmentation de la concentration d'ATP à 4 heures d'incubation dans les
tranches incubées en absence de CAA par rapport à celles mesurées à 1 et à 2
heures d'incubation sans que nous ayons d'interprétation pour expliquer ce
phénomène. Malgré la diminution de 26% de la concentration d'ATP mesurée après
24 heures d'incubation dans les conditions témoin sans CAA (par rapport aux
concentrations d'ATP mesurées après 1 heure d'incubation), une valeur d'ATP de
6.2 µmol/g protéines est satisfaisante et montre une bonne viabilité de nos tranches
après 24 heures d'incubation.
Parmi les paramètres étudiés du métabolisme du lactate, la néoglucogenèse est le
plus sensible à l'action du CAA. A partir de deux heures d'incubation, la synthèse du
glucose est inhibée de manière significative. A 4 heures, nous trouvons une inhibition
de 50% par rapport au contrôle sans CAA. L'utilisation du lactate et l'accumulation du
pyruvate sont moins sensibles que la néoglucogenèse. Elles sont inhibées au-delà
de 4 heures et de 16 heures d'incubation, respectivement.
En regardant le profil de la néoglucogenèse au cours du temps, en absence de CAA,
nous constatons que le glucose qui a été formé pendant les premières heures
d'incubation est utilisé au delà. La synthèse du glucose atteint son niveau maximum
après 6 heures d'incubation. Au bout de 24 heures on ne trouve que 62% du glucose
mesuré à 6 heures (99.4 ± 17.3 vs. 160.9 ± 13.4 µmol/g protéines, respectivement).
Cela suppose que la voie de la néoglucogenèse était prépondérante au début de
l'incubation, et qu'au delà de 6 heures, la glycolyse est devenue plus importante par
rapport à la néoglucogenèse. Il est à noter que la quantité mesurée de glucose
représente la somme des deux voies métaboliques (la synthèse du glucose et la
En fait, malgré la prédominance des segments tubulaires néoglucogéniques, d'autres
segments des néphrons ayant une activité glycolytique peuvent exister dans le
cortex rénal comme la branche épaisse ascendante de l'anse de Henlé et le tubule
contourné distal [246]. Il a été montré que ces segments contiennent les enzymes
clés de la glycolyse (HK, PFK et PK) [247]. Ce sont probablement ces segments
glycolytiques qui sont responsables de l'utilisation du glucose observée au-delà de 6
heures d'incubation. Il faut probablement atteindre une certaine concentration de
glucose dans le milieu d'incubation avant que ce dernier puisse pénétrer dans les
cellules glycolytiques, où il sera utilisé.
En outre, nous trouvons, en présence de 75 µM CAA, un profil de la néoglucogenèse
qui ressemble à celui observé en absence de CAA au cours du temps ; néanmoins,
l'utilisation du glucose n'est qu'apparente car sa concentration n'est pas
statistiquement différente entre 24 heures (52.1 ± 7.5 µmol/g protéines) et 8 heures
d'incubation (maximum du glucose : 72.2 ± 5.1 µmol/g protéines) en présence du
toxique. L'absence de l'utilisation du glucose en présence de 75 µM CAA signifie que
le CAA exerce son effet inhibiteur non seulement sur la voie de la néoglucogenèse,
comme le montrent les résultats ci-dessus, mais certainement aussi sur la voie de la
glycolyse, probablement via l'inhibition induite par le CAA de la GAPDH (Tableau 2,
page 87). L'inhibition de l'activité GAPDH sera discutée ultérieurement.
L'inhibition de la néoglucogenèse, observée en présence du CAA, peut être la
conséquence de l'inhibition de certaines activités enzymatiques de cette voie
métabolique, ou de la diminution d'ATP cellulaire provoquée par le CAA, l'ATP étant
nécessaire pour la synthèse du glucose. Une inhibition de la chaîne respiratoire
mitochondriale pourrait être responsable de la diminution d'ATP.
Nos résultats montrent que la néoglucogenèse est la voie métabolique la plus
sensible à l'effet du CAA. C'est pour cette raison que certaines activités
enzymatiques de cette voie ont été mesurées. Sont mesurées également la
respiration mitochondriale et l'oxydation du lactate marqué. Le temps de 4 heures
d'incubation est choisi pour réaliser la suite de cette partie car l'effet de 75 µM CAA
est observé, à l'exception de l'accumulation du pyruvate, sur tous les paramètres
étudiés à partir de ce temps là.
2-3. EFFET DE 75 µM CAA SUR L'ACTIVITE DE CERTAINES
ENZYMES DE LA NEOGLUCOGENESE DANS DES TRANCHES DE
Nous avons étudié l'effet de 75 µM CAA sur l'activité de cinq enzymes impliquées
dans la synthèse du glucose, dont les trois activités clés qui catalysent les réactions
irréversibles de cette voie (PEPCK, F1,6-BPase et G6-Pase), la LDH qui initie la voie
néoglucogénigue à partir du lactate et enfin la GAPDH, l'enzyme impliquée dans la
néoglucogenèse et la glycolyse et qui contient des groupes thiols dans son site actif.
Les activités GAPDH, F1,6-BPase et G6-Pase sont inhibées par le CAA tandis que
les activités LDH et PEPCK ne le sont pas. L'inhibition de ces activités par le CAA
pourrait être expliquée par la capacité du CAA à se lier aux composés thiols [113,
116], induisant ainsi la diminution de ces composés. Dans cette étude, nous avons
montré une diminution de 66% des thiols totaux mesurés dans des tranches de
cortex rénal incubées pendant 4 heures en présence de 75 µM CAA, par rapport au
contrôle sans CAA (2.6 vs. 7.7 µmol/g protéines, respectivement). Des composés
cystéines font partie de manière plus ou moins importante des sites actifs des
enzymes de la néoglucogenèse que nous avons étudiées.
La GAPDH est connue pour avoir trois groupes thiols, dont un essentiel, dans son
site actif [248, 249]. Le groupe thiol essentiel de cette enzyme réagit avec
l'iodoacétate, un agent sulfhydryl qui réagit avec les résidus cystine, ce qui affaiblit la
fixation de NAD+ à la GAPDH et inhibe l'enzyme [248]. L'inhibition de la GAPDH par
l'iodoacétate est également observée par Wolf et al. [250]. De même, l'acide
hypochlorique (HOCl), qui induit une baisse du glutathion et des groupes thiols,
inhibe l'activité GAPDH [251].
La F1,6-BPase est aussi caractérisée par la présence d'un groupe sulfhydryl
hautement réactif et nécessaire pour son fonctionnement [252]. Le traitement de
cette enzyme par la N-éthylmaléimide (NEM), un composé sulfhydryl réactif dérivé
de l'acide maléique et utilisé pour modifier les résidus cystine, conduit à une
modification de ce groupe. Ceci conduit, par conséquent, à une modification des
propriétés de cette enzyme, notamment une perte de l'inhibition de l'enzyme par le
F2,6-P2 [253], une absence de l'activation de l'enzyme par les cations monovalents
[254] et une perte de 60% de l'activité catalytique de l'enzyme [255]. En ce qui
concerne la G6-Pase, trois groupes thiols, dont un se trouvant dans sa sous unité
catalytique, sont importants pour son activité [256, 257].
Contrairement à ces trois enzymes, et malgré la présence de groupes thiols dans la
composition de la LDH, il semble que ces groupes ne sont pas essentiels pour le
fonctionnement de l'enzyme [251, 258]. L'activité LDH ne change pas sous l'effet
d'acide hypochlorique (HOCl) malgré la diminution du glutathion et des groupes
thiols induite par l'acide [251].
Il en est de même pour la PEPCK, dont l'activité n'est pas inhibée par le CAA,
malgré la présence de groupes sulfhydryls et des résidus cystine importants dans
son site catalytique [259]. Les nucléotides, qui fournissent une protection complète
contre l'inactivation de la PEPCK, pourraient être présents en quantités optimales
dans les cellules intactes, ce qui expliquerait l'absence d'inhibition de la PEPCK par
En outre, d'autres activités enzymatiques sont sensibles à la diminution des fonctions
SH. Dans une étude réalisée chez la souris par Soares et al., l'exercice physique
associé à un régime alimentaire sans sélénium, qui constitue le centre de la
glutathion peroxydase, peut inhiber des enzymes contenant des groupes sulfhydryls
comme l'alanine déshydrogénase et la succinate déshydrogénase, mais pas la LDH.
Cette inhibition peut être attribuée à l'oxydation des groupes thiols par les molécules
oxygénées réactives (ROS) produits par l'exercice physique [258].
En conclusion, l'importante diminution des thiols totaux, qui implique la diminution
déjà observée du glutathion, explique l'inhibition d'activité de certaines enzymes
contenant des groupes thiols essentiels pour leur fonctionnement et confirme les
observations des autres auteurs montrés ci-dessus. La conséquence logique de
l'inhibition de certaines activités enzymatiques d'une voie métabolique est l'inhibition
de cette voie. La néoglucogenèse est donc inhibée. Il est évident dans nos résultats
que l'activité GAPDH est la plus sensible à l'action du CAA. Elle est inhibée de 53%
en présence de 75 µM CAA comparée au contrôle sans CAA. La GAPDH catalyse
une réaction réversible et donc considérée comme une enzyme à la fois de la
néoglucogenèse et de la glycolyse. La forte inhibition de l'activité GAPDH pourrait
expliquer l'inhibition de ces deux voies métaboliques en présence de 75 µM CAA et,
par conséquent, l'absence de l'utilisation du glucose en présence du toxique, alors
que la glycolyse était active dans les conditions témoin sans CAA.
2-4. EFFET DE 75 µM CAA SUR LA FONCTION MITOCHONDRIALE
ET L'OXYDATION DU LACTATE DANS DES TRANCHES DE CORTEX
Nos résultats montrent que 75 µM CAA diminue de 35% la concentration d'ATP
cellulaire à 4 heures d'incubation par rapport au contrôle sans CAA. Pour expliquer
cette diminution, nous avons étudié l'effet de cette concentration de CAA sur la
consommation d'oxygène dans des mitochondries isolées de tranches de cortex
rénal. La phosphorylation oxydative est la source principale d'ATP dans les cellules
oxydatives. Nous avons également étudié l'effet de 75 µM CAA sur la formation de
[14CO2] à partir de [U-14C]-lactate par ces tranches. Nous avons trouvé que le CAA
inhibe le complexe I, mais pas le complexe II de la chaîne respiratoire mitochondriale.
L'absence d'inhibition du complexe II implique l'intégrité des complexes III et IV de la
chaîne respiratoire. L'inhibition du complexe I était comprise entre 22% et 23% par
rapport au contrôle sans CAA, pour les différents donneurs d'électrons utilisés
(mélange malate + pyruvate ou mélange malate + glutamate, respectivement). En
mesurant la formation du [14CO2] résultant de l'oxydation complète de [U-14C]-lactate,
nos avons trouvé une inhibition de 19% en présence de 75 µM CAA comparé au
contrôle sans CAA, ce qui est du même ordre de grandeur que l'inhibition du
Nos résultats confirment ceux de Nissim et al. [43] qui ont trouvé que le traitement
par l'IFO inhibe la phosphorylation oxydative au niveau du complexe I de la chaîne
respiratoire. Cette inhibition était associée à une augmentation du NADH, une
diminution du NAD+ et une diminution du flux enzymatique à travers la PDH et le
cycle des acides tricarboxyliques. Leur étude in vitro avait montré que le CAA est le
métabolite de l'IFO responsable de l'inhibition du complexe I. Il est à noter que nous
avons obtenu l'inhibition du complexe I en utilisant une concentration faible de CAA
(75 µM) par rapport à celle que Nissim et al. avaient utilisée (1000 µM). La
concentration de CAA que nous avons utilisée est proche de celles mesurées chez
des patients traités par l'IFO [33, 96, 109-111]. Il faut également préciser que Nissim
et al. avaient appliqué le CAA directement sur des mitochondries isolées de tissu
sain, tandis que, dans nos expériences, le toxique est appliqué sur des tranches de
cortex rénal pendant 4 heures avant l'extraction des mitochondries. La chute
complète d'ATP cellulaire à 24 heures d'incubation en présence de 75 µM CAA
signifie probablement une inhibition totale de la phosphorylation oxydative entraînant
la nécrose cellulaire.
Il est intéressant de savoir que le complexe I de la chaîne respiratoire contient huit
résidus cystine ayant un rôle important dans sa structure et sa fonction [260]. Ceci
donnerait une explication au mécanisme d'inhibition du complexe I par le CAA. En
effet, il a été montré que l'incubation des tranches de cerveau de souris avec
l'iodoacétate conduit à une inhibition du complexe I qui donc est sensible aux agents
qui modifient les groupes thiols. L'ajout du glutathion ou des thiols antioxydants a pu
inverser cette inhibition ce qui signifie que l'oxydation des groupes thiols dans le
complexe I serait responsable de la diminution de l'activité de ce complexe [261,
262]. Dans cette étude, nous avons observé une diminution des thiols totaux sous
l'effet de 75 µM CAA. Cette diminution pourrait donc expliquer l'inhibition du
complexe I de la chaîne respiratoire.
Cette étude montre que le mécanisme moléculaire de la néphrotoxicité du CAA
implique une déplétion du glutathion ainsi que des thiols totaux et une toxicité
mitochondriale se manifestant par la diminution d'ATP cellulaire. Un autre
mécanisme de la toxicité rénale du CAA pourrait être proposé. Il s'agit du stress
oxydatif et de l'augmentation de la péroxydation des lipides. Le malondialdéhyde
(MDA) est un indice de la péroxydation des lipides. Il a été montré dans une étude
préalablement réalisée dans notre laboratoire, que l'incubation des tubules rénaux
proximaux isolés pendant 4 heures avec 1 mM lactate dans le tampon Krebs-
Henseleit en présence de 75 µM CAA, avait induit une augmentation de 66% du
MDA par rapport au contrôle sans CAA. Ces résultats confirment ceux obtenus par
Springate et al., qui avaient montré que la toxicité rénale du CAA in vivo était
associée à une déplétion sévère du glutathion et une accumulation du MDA [80], et
confirment également les résultats in vivo de Sehrili et al. qui ont trouvé que le
traitement des rats par l'IFO diminue le glutathion et augmente le MDA [263]. Le
stress oxydatif altère aussi la fonction et la viabilité des cellules tubulaires rénales
proximales en culture en diminuant l'ATP cellulaire et en augmentant la péroxydation
des lipides [264, 265]. Dans nos expériences, l'inhibition du complexe I de la chaîne
respiratoire peut engendrer un stress oxydatif qui est accompagné d'une
augmentation des molécules oxygénées réactives (ROS) expliquant l'augmentation
de MDA en présence de CAA.
Dans cette première partie, nous avons montré que le CAA, utilisé à des
concentrations faibles proches de celles mesurées chez les patients traités par l'IFO,
induit un effet toxique sur les tranches de cortex rénal. La toxicité du CAA s'est
manifestée par une inhibition de la phosphorylation oxydative dans les mitochondries
ainsi qu'une inhibition du métabolisme du lactate et de la néoglucogenèse résultant
de l'inhibition de certaines activités enzymatiques de cette voie, notamment la
GAPDH. L'inhibition par le CAA de la GAPDH, qui est à la fois une enzyme de la
néoglucogenèse et de la glycolyse, pourrait provoquer une inhibition de ces deux
voies métaboliques. L'inhibition de la GAPDH par l'acide koningique, par exemple,
avait tué des cellules ayant une activité glycolytique élevée via une déplétion glucose
dépendante d'ATP [266]. Etant donné que les cellules cancéreuses exhibent un taux
élevé de glycolyse même en présence d'oxygène (effet Warburg), nous avons voulu
savoir si le CAA pouvait, en inhibant cette voie, altérer la croissance des cellules
cancéreuses et, par conséquent, participer à l'effet thérapeutique de l'IFO.
L'inhibition de la glycolyse a été proposée par plusieurs auteurs comme une stratégie
de traitement du cancer [129, 180-182, 184, 185]. Contrairement à son rôle dans la
toxicité rénale de l'IFO, un rôle anticancéreux éventuel du CAA a été peu étudié.
L'objectif de la deuxième partie est donc de rechercher cet effet éventuel. Elle sera
réalisée sur les cellules de cancer du sein MCF-7 qui sont strictement dépendantes
de la présence du glucose dans le milieu de culture pour leur croissance,
contrairement à d'autres types des cellules cancéreuses [267].
3. DEUXIEME PARTIE : EFFET DU CAA SUR LES CELLULES
DE CANCER DU SEIN (MCF-7)
3-1. EFFET DU CAA SUR LA PROLIFERATION CELLULAIRE ET LE
METABOLISME DU GLUCOSE
Dans cette deuxième partie nous avons étudié l'effet du CAA sur les cellules de
cancer du sein, MCF-7, l'objectif étant de rechercher une contribution éventuelle du
CAA à l'effet thérapeutique de l'IFO. Nous avons choisi les cellules MCF-7 car l'IFO
est utilisé dans plusieurs protocoles de traitement de cancer du sein avancé et
résistant [22]. Il s'agit également d'une souche de cellules exclusivement dépendante
du glucose pour sa croissance et donc adaptée à une étude sur une éventuelle
inhibition de la voie de la glycolyse [267].
Les concentrations de CAA choisies pour la deuxième partie sont, comme pour la
première partie, proches de celles mesurées chez les patients traités par l'IFO (entre
5 et 200 µM) [33, 96, 109-111]. Elles étaient comprises entre 0 et 50 µM. Les cellules
ont été incubées pendant 24 heures dans le milieu RPMI-1640 contenant 2 g/l de
glucose et 5% de SVF. Nous avons étudié la prolifération cellulaire en déterminant le
nombre total et la viabilité des cellules et en étudiant la morphologie des cellules à
l'aide d'un microscope lié à un ordinateur et un logiciel.
Nos résultats montrent clairement que le CAA inhibe la prolifération des cellules
MCF-7 de manière dose dépendante. En absence de CAA, le nombre des cellules
augmente de 45% pendant les 24 heures d'incubation (1.31 vs. 1.88 x106
cellules/puits). La présence de 10 µM CAA inhibe la prolifération cellulaire (19%
d'augmentation du nombre des cellules par rapport aux cellules non incubées). La
prolifération est complètement abolie par 25 µM CAA (1.26 vs. 1.31 x106 cellules
/puits avant l'incubation). Le nombre des cellules vivantes, déterminé par le
cytomètre, représente 80% du nombre total des cellules. La viabilité est également
montrée par la forte teneur en ATP cellulaire. Les photos des cellules confirment ces
résultats. Elles montrent l'inhibition dose dépendante de la prolifération en présence
de CAA. La morphologie des cellules montre qu'elles sont vivantes en présence de
25 µM CAA malgré l'arrêt de la croissance.
Dans ce contexte, il est à noter que la présence de 50 µM CAA a conduit à une
destruction quasi totale des cellules. Les rares cellules qui restent deviennent rondes
et flottent dans le milieu. Par conséquent, les dosages des différents paramètres
étaient dénués de sens en présence de cette concentration.
Depuis les travaux de Warburg, il est communément admis que les cellules
cancéreuses sont caractérisées par un taux élevé de glycolyse même en présence
d'oxygène [123]. La glycolyse utilise le glucose pour produire du lactate même en
présence d'oxygène (effet Warburg). L'utilisation du glucose par les cellules
cancéreuses est généralement associée à la production d'énergie. L'inhibition de la
glycolyse et de la voie des pentoses phosphate est proposée comme une stratégie
pour inhiber la prolifération des cellules cancéreuses, de manière préférentielle
(Figure 9, page 48). Voici quelques exemples : le 3-bromopyruvate et la lonidamine
sont proposés comme inhibiteurs de l'HK [191, 193, 268], l'imatinib inhibe l'activité
HK et G6PDH [202], l'oxythiamine inhibe la voie des pentoses phosphate via
l'inhibition des transkétolases [200] et enfin, l'acide acétylsalicylique et l'acide
salicylique inhibent l'activité PFK [185]. Nous avons donc choisi d'étudier l'effet du
CAA sur la voie de la glycolyse et sur la production d'énergie (ATP cellulaire) dans
les cellules MCF-7.
Nos résultats montrent une inhibition de l'utilisation du glucose par les cellules MCF-
7 en présence de 10 et 25 µM CAA. Cette inhibition est accompagnée d'une
inhibition de la production du lactate en présence de 25 µM CAA. Au vu de cette
inhibition et de l'importance du métabolisme du glucose dans la production d'énergie
dans les cellules cancéreuses, une diminution de l'ATP cellulaire était logiquement
attendue. D'une manière surprenante mais intéressante, l'ATP cellulaire n'était pas
inhibé par les concentrations utilisées de CAA. Au contraire, il a augmenté de 53%
en présence de 25 µM CAA comparé au contrôle sans CAA. Ces résultats montrent
qu'un inhibiteur de la glycolyse pourrait induire une inhibition de la prolifération des
cellules cancéreuses sans diminuer leur contenu d'ATP, contrairement à ce qui a été
déjà montré par l'effet du 3-bromopyruvate [190, 192]. Nous pouvons nous référer
également aux travaux de Lee et al. qui ont montré que la privation des cellules
MCF-7 du glucose induit l'apoptose cellulaire accompagnée d'une diminution d'ATP
cellulaire [269].
Ces résultats suggèrent également que la glycolyse ne soit pas la source unique de
la production d'ATP car l'inhibition d'environ 45% de cette voie métabolique n'induit
pas une diminution d'ATP en présence de 25 µM CAA. L'ATP pourrait donc être
produit par d'autres sources, notamment oxydative.
En effet, le ratio lactate produit/glucose utilisé est compris entre 1.36 et 1.52 en
absence et en présence de CAA, respectivement. Cela veut dire que le pourcentage
du glucose transformé en lactate représente, au maximum, 75% du glucose utilisé.
Donc, une partie de ce glucose utilisé pourrait être oxydé dans le cycle des acides
tricarboxyliques. Effectivement, et comme le montrent le Tableau 7, page 97, environ
6% du glucose utilisé est complètement oxydé en [14CO2] en présence ou en
absence de 25 µM CAA. Il est utile de rappeler que l'oxydation complète d'un
glucose produit 36 molécules d'ATP tandis que le métabolisme d'un glucose par la
voie glycolytique ne produit que 2 ATP. Si seulement une petite partie du glucose
consommé était oxydée, elle contribuerait de manière significative à la production
d'ATP. Guppy et al. [270] avaient montré dans les cellules MCF-7 que la production
d'ATP était 80% oxydative et 20% glycolytique. Les contributions de la partie
oxydative étaient : 10% glucose, 14% glutamine, 7% palmitate, 4% oléate et 65% de
sources non identifiées. Ainsi, l'ATP provenant du glucose (voie glycolytique et voie
oxydative) et de la glutamine représentait 28.8% et 10.7% de l'ATP total,
respectivement. Pour Guppy et al. le métabolisme du glucose et de la glutamine
contribue donc à 40% (moins de la moitié) du turnover d'ATP.
3-2. MECANISME D'AUGMENTATION D'ATP CELLULAIRE EN
La prolifération cellulaire consomme une quantité importante d'énergie. Nos résultats
montrent que cette prolifération est complètement abolie en présence de 25 µM CAA,
concentration de CAA en présence de laquelle l'ATP augmente. L'absence de
prolifération cellulaire, et donc l'absence d'utilisation d'ATP, pourraient contribuer à
l'augmentation de ce dernier avec 25 µM CAA.
Il est à noter que la concentration de 50 µM CAA avait conduit à une destruction
quasi totale des cellules après 24 heures d'incubation. Il était donc dénué de sens de
doser l'ATP cellulaire en présence de cette concentration. Il est probable que les
fortes concentrations de CAA exercent d'autres effets sur les cellules cancéreuses,
probablement au niveau de la chaîne respiratoire, conduisant à la chute d'ATP et à la
nécrose cellulaire. Cette possibilité est proposée par Bruggemann et al. [213] et
Motrescu et al. [214].
3-3. MECANISME D'INHIBITION DE LA PROLIFERATION
CELLULAIRE EN PRESENCE DE CAA
Devant l'absence de la diminution d'ATP en présence de CAA, nous nous sommes
posés la question de savoir le mécanisme par lequel la prolifération cellulaire est
inhibée en présence de 10 et 25 µM CAA.
La glycolyse et la voie des pentoses phosphate, initiée par la G6PDH à partir du G6-
P, fournissent de nombreux métabolites nécessaires pour la biosynthèse [145]. Par
exemple : le ribose-5-phosphate est produit par le cycle des pentoses phosphate par
voie oxydative ou non oxydative [271]. D'autres intermédiaires comme le DHAP (le
précurseur des phospholipides et des triacylglycérols) ou le 3-PG (indispensable
pour la synthèse de la sérine, la cystéine et la glycine) sont des éléments importants
dans la prolifération cellulaire [272].
Nous avons montré que la quantité du glucose complètement oxydé en [14CO2]
représente environ 6% de la quantité totale du glucose utilisé (Tableau 7, page 97),
soit 0.28 µmol/106 cellules en absence de CAA et 0.15 µmol/106 cellules en présence
de 25 µM CAA (les valeurs du glucose consommé sont 4.6 et 2.6 µmol/106 cellules
en absence et en présence de 25 µM CAA, respectivement, voir Figure 24, page 94).
Supposons que tout le lactate produit par nos cellules provienne du métabolisme du
glucose, le calcul suivant donne les valeurs de la quantité du glucose éventuellement
disponible pour la biosynthèse. En soustrayant la quantité de lactate produit (1
lactate = ½ glucose) et la quantité du glucose complètement oxydé en [14CO2] de la
quantité totale du glucose consommé, nous trouvons que le glucose disponible pour
la biosynthèse diminue de 1.19 µmol/106 cellules en absence de CAA à 0.46
µmol/106 cellules en présence de 25 µM CAA (voir Figures 24 et 25, pages 94 et 95).
Une inhibition de la glycolyse en présence de 10 et 25 µM CAA, comme le montrent
nos résultats, pourrait donc diminuer la provision des précurseurs des biosynthèses,
ce qui pourrait avoir des répercussions importantes sur la prolifération des cellules
cancéreuses caractérisées par un taux élevé de croissance.
Dans une très récente publication de la revue Science, Vander Heiden et al. [147]
ont proposé que l'effet Warburg soit beaucoup plus lié à la formation de précurseurs
de la biosynthèse et à celle de NADPH qu'à la formation d'énergie. En effet, les
cellules proliférantes doivent synthétiser tout leur contenu cellulaire. Si l'ATP est
effectivement nécessaire à ces biosynthèses, les précurseurs qui sont fournis
presque exclusivement par le glucose et par la glutamine le sont tout autant. D'après
ces auteurs, l'acétyl CoA nécessaire à la synthèse du palmitate (un constituant
majeur des membranes cellulaires) provient exclusivement du glucose. Or, la
quantité d'ATP produite par le métabolisme du glucose jusqu'à la formation d'acétyl
CoA est de 8 moles pour un glucose consommé (et donc 32 ATP pour 4 glucose),
alors que le NADPH produit par la voie des pentoses phosphate est de 2 moles pour
un glucose (8 NADPH pour 4 glucose). Dans le même temps, la synthèse d'un acide
palmitique nécessite l'apport de 7 ATP et de 14 NADPH (Figure 31). La
transformation de glucose en acétyl CoA fournit donc 4 fois plus d'ATP que de
NADPH alors que la synthèse d'un acide palmitique utilisera 2 fois plus de NADPH
que d'ATP. Il est donc nécessaire pour les cellules en croissance d'utiliser de fortes
quantités de glucose même en présence d'O2 (effet Warburg) afin de former les
précurseurs indispensables aux biosynthèses. De plus, les cellules proliférantes ont
toujours des rapports ATP/ADP élevés, ce qui semble signifier que l'énergie n'est
pas un facteur limitant dans la prolifération. Ces propositions sont tout à fait en
accord avec nos résultats puisque nous relions l'inhibition de la prolifération cellulaire
à l'inhibition de la glycolyse induite par le CAA, et ainsi à la diminution des
précurseurs nécessaires aux voies de synthèse, et non pas à la chute d'ATP
Figure 31et ainsi à la diminution des précurseurs nécessaires aux voies de synthèse,
et non pas à la chute d'ATP cellulaire.
Figure 32 : Le rôle de la glycolyse dans la formation du palmitate.
4 Fructose 1,6-Bis-P
8 Glyceraldéhyde 3-P
8 1,3-Bis-P Glycérate
3-4. EFFET DU CAA SUR LES ACTIVITES ENZYMATIQUES DE LA
GLYCOLYSE ET SUR LE TRANSPORT CELLULAIRE DU GLUCOSE.
MECANISME D'INHIBITION DE L'UTILISATION DU GLUCOSE EN
Pour expliquer l'inhibition observée de la consommation du glucose par les cellules
MCF-7 nous avons mesuré les activités de certaines enzymes impliquées dans la
régulation de la voie de la glycolyse et de la voie des pentoses phosphate (Figure 4,
page 19). Nous avons également étudié le transport cellulaire du glucose en utilisant
le DOG, un analogue non métabolisable du glucose.
L'activité PFK, l'enzyme qui catalyse la réaction régulatrice principale de la glycolyse,
n'est pas modifiée en présence de CAA. Les activités HK et PK diminuent de 29% et
de 26%, respectivement en présence de 25 µM CAA. La GAPDH est à la fois une
enzyme de la glycolyse et de la néoglucogenèse. Comme nous l'avons montré dans
la première partie, son activité est inhibée de 53% dans les tranches de cortex rénal
en présence de 75 µM CAA. Dans les cellules MCF-7, l'activité GAPDH diminue de
18 et 62% en présence de 10 et 25 µM CAA, respectivement. Il faut rappeler que la
GAPDH contient des groupes thiols dans son site actif [248, 249]. Dubourg et al.
avaient montré que le CAA peut se lier aux composés thiols [113], ce qui pourrait
expliquer l'inhibition de cette enzyme.
Parmi les différentes activités enzymatiques mesurées, il semble que l'HK catalyse la
réaction limitante de la glycolyse car son activité est la plus faible de toutes celles
mesurées dans les conditions témoins (12.0 mUI/106 cellules vs. 43.0 mUI/106
cellules pour la PFK et 412.5 mUI/106 cellules pour la PK). Cette activité diminue de
29% en présence de 25 µM CAA. Malgré la diminution importante observée de
l'activité GAPDH en présence de 25 µM CAA, il est peu probable qu'elle soit
responsable de la diminution de la glycolyse observée dans notre étude. En effet,
aucune accumulation de GAP, le substrat de la réaction catalysée par la GAPDH,
n'est détectée ni en présence ni en absence de CAA (n = 2). Le DHAP et le F1,6-P2,
deux autres métabolites de la voie glycolytique, ont été également dosés et ils
étaient non détectables dans toutes les conditions expérimentales (n = 2).
Contrairement aux autres activités enzymatiques qui ont diminué ou qui n'ont pas
changé, l'activité G6PDH est la seule qui augmente en présence de 5 et 10 µM CAA.
Cette augmentation pourrait être expliquée par la précédente démonstration que le
CAA induit un stress oxydatif [80, 121] ainsi que par les observations d'Ursini et al.
qui ont montré une surexpression de la G6PDH dans des différents types cellulaires
humains après traitement par des agents qui modifient les voies biochimiques
responsables du statut des composés redox intracellulaires. Ceci pourrait
représenter un mécanisme de protection contre le stress oxydatif [273]. Il est
également utile de mentionner une étude préalablement réalisée dans notre
laboratoire où 75 µM CAA avait induit une augmentation du MDA (un indice du stress
oxydatif) dans des tubules rénaux proximaux isolés du rat (voir page 119). Bien que
les paramètres du stress oxydatif n'aient pas été mesurés dans cette étude, il est fort
probable que les concentrations utilisées de CAA l'ont provoqué dans les cellules
MCF-7 induisant une augmentation de l'expression de l'activité G6PDH.
Le transport cellulaire du glucose est une étape essentielle afin d'initier son
métabolisme. Les cellules cancéreuses sont caractérisées par un taux élevé de
captation du glucose [167]. Les transporteurs du glucose sont surexprimés dans les
cellules de cancer du sein [168, 169]. Il a été montré que l'inhibition du transport du
glucose, par la naringénine par exemple, inhibe la prolifération des cellules MCF-7
[205]. Le transport du glucose était linéaire entre 5 et 35 minutes dans une étude
réalisée par Singh et al. sur les cellules MCF-7 [209]. Nous avons donc choisi
d'étudier le transport de DOG pendant une période de 35 minutes en absence ou en
présence de 5, 10 et 25 µM CAA. Nos résultats montrent clairement une inhibition de
la captation du DOG de manière dose dépendante et significative avec toutes les
concentrations utilisées (17%, 32 et 33% d'inhibition en présence de 5, 10 et 25 µM
CAA, respectivement par rapport au contrôle sans CAA).
Il est évident que le transport cellulaire du glucose est plus sensible à l'effet du CAA
que les activités enzymatiques de la glycolyse puisque le transport est inhibé par des
concentrations plus faibles du CAA (5 et 10 µM) qui ne modifient pas l'activité de ces
enzymes. L'inhibition du transport du glucose semble être le premier événement
dans le mécanisme d'effet du CAA sur les cellules MCF-7. En conjonction avec
l'inhibition de l'activité HK, l'inhibition du transport du glucose sous l'effet du CAA
pourrait expliquer l'inhibition du métabolisme du glucose (la diminution de la
consommation du glucose et de la production du lactate) et par conséquent,
l'inhibition de la prolifération cellulaire.
3-5. ANALOGIE DE STRUCTURE ET D'EFFETS ENTRE LE CAA ET
LE 3-BROMOPYRUVATE
Le 3-bromopyruvate a été décrit comme une molécule pouvant être efficace vis-à-vis
des cellules cancéreuses. Cette molécule possède un halogène (brome) porté par un
carbone en position α par rapport
à une fonction cétonique : Br-CH2-CO-COO-. Le
CAA possède également dans sa structure un halogène (chlore) porté par un
carbone en position α par rapport à une fonction aldéhyde : Cl-CH2-CHO. L'analogie
de structure est évidente entre ces deux composés. Le 3-bromopyruvate tout comme
le CAA est une molécule susceptible de se lier aux composés possédant des
groupements thiols dans leur structure [115, 274]. L'effet du 3-bromopyruvate sur les
cellules cancéreuses provient d'une diminution de la glycolyse par inhibition de
l'activité HK-II particulièrement active dans ces cellules [156, 157]. Au cours de cette
étude, nous avons montré que le CAA inhibe la glycolyse et que l'activité HK est
diminuée par ce composé. Enfin, l'action du 3-bromopyruvate provoque une chute
d'ATP cellulaire probablement en liaison avec une diminution de la phosphorylation
oxydative [189-191]. Bien que nous n'ayons pas mesuré dans cette étude l'effet du
CAA sur la chaîne respiratoire, la destruction quasi totale des cellules observée pour
la concentration de 50 µM CAA pourrait être la conséquence d'une chute d'ATP
engendrée par une inhibition de la chaîne respiratoire mitochondriale. Nos résultats
suggèrent que le CAA pourrait agir par un mécanisme analogue à celui décrit pour le
3-bromopyruvate. Cette hypothèse doit être confirmée par des études menées en
parallèle avec les deux toxiques sur les cellules MCF-7 et sur d'autres types de
cellules cancéreuses pour voir si ce mécanisme d'action des toxiques est
comparable et peut être généralisé à d'autres types de cancer.
Dans cette partie, nous avons démontré que le CAA inhibe la prolifération des
cellules de cancer du sein, MCF-7. Cet effet inhibiteur du CAA est observé en
présence de concentrations faibles du toxique, proches de celle trouvées dans le
sang des patients traités par l'IFO. Nos résultats suggèrent que le CAA ne soit pas
impliqué uniquement dans la néphrotoxicité de l'IFO, mais qu'il pourrait contribuer à
son effet thérapeutique. L'IFO possède une activité anticancéreuse plus importante
que le CP dans certaines tumeurs humaines [101] ou expérimentales [211], et il peut
être efficace dans les cas d'échec du traitement par le CP. Cela pourrait être
expliqué par le fait que le métabolisme hépatique de l'IFO produit une quantité plus
élevée de CAA que le métabolisme du CP en raison de la différence structurelle
entre les deux isomères.
Nos résultats montrent également que l'inhibition de la glycolyse, suite à l'inhibition
du transport du glucose et de l'activité HK, et par conséquent la diminution des
provisions de précurseurs des biosynthèses, est probablement à l'origine de
l'inhibition de la prolifération cellulaire, qui n'est pas associée à une diminution d'ATP
cellulaire. La glycolyse n'est donc pas la source unique d'énergie dans les cellules
MCF-7, car l'inhibition de 45% de cette voie métabolique par 25 µM CAA n'est pas
associé à une diminution d'ATP. Nous avons trouvé que l'oxydation du glucose
représente environ 6% du glucose consommé. La phosphorylation oxydative dans
les mitochondries pourrait donc participer, de manière significative, à la production
Pour compléter notre l'étude, nous avons réalisé des cultures des cellules MCF-7 en
conditions d'hypoxie afin d'étudier le métabolisme du glucose dans ces conditions.
Nous avons émis l'hypothèse que l'hypoxie, en inhibant la phosphorylation oxydative,
pourrait inhiber la production d'ATP par la chaîne respiratoire.
3-7. EFFET DU CAA SUR LES CELLULES MCF-7 EN HYPOXIE
L'hypoxie est une caractéristique importante du microenvironnement tumoral,
notamment dans les tumeurs solides, agressives, résistantes au traitement et les
tumeurs ayant un mauvais pronostic [275]. Nous avons voulu savoir si l'hypoxie
renforcerait l'effet du CAA sur les cellules MCF-7 en inhibant la phosphorylation
oxydative, qui représente une source probablement importante d'ATP de ces cellules.
Pour cela, nous avons étudié l'effet d'hypoxie (5% O2) sur la prolifération et l'énergie
cellulaires, et sur le métabolisme du glucose dans les cellules MCF-7.
Comparativement aux conditions de normoxie, l'hypoxie n'a pas inhibé la
prolifération des cellules MCF-7 après 24 heures d'incubation (1.82 x106
cellules/puits en hypoxie vs. 1.88 x106 cellules/puits en normoxie). En présence de
CAA, le profil d'effet du CAA sur la prolifération cellulaire n'est pas modifié en
hypoxie par rapport à la normoxie : inhibition identique de la prolifération en présence
de 10 et 25 µM CAA. Sur le métabolisme du glucose, le CAA induit le même profil
d'effet en hypoxie qu'en normoxie : inhibition de la consommation du glucose en
présence de 10 et 25 µM CAA, inhibition de la production du lactate en présence de
25 µM CAA, inhibition des activités HK, PK, GAPDH en présence de 25 µM CAA,
augmentation de l'activité G6PDH avec 5 et 10 µM CAA, et absence d'effet sur
l'activité PFK, par rapport aux conditions témoin sans CAA.
Il est évident que l'hypoxie appliquée dans nos expériences a conduit, dans le
contrôle sans CAA, à une augmentation de près de 100% de l'utilisation du glucose
et de la production du lactate, par rapport aux conditions de normoxie (8.3 vs. 4.6
µmol/106 cellules et 13.5 vs. 6.3 µmol/106 cellules, respectivement). L'hypoxie a
également induit, dans le contrôle sans CAA, une surexpression de toutes les
activités enzymatiques étudiées, sauf la G6PDH, comparativement à la normoxie.
En ce qui concerne l'énergie cellulaire, le profil d'effet du CAA sur l'ATP n'est pas
modifié en hypoxie par rapport à la normoxie (pas d'effet sur l'ATP avec 5 et 10 µM
CAA, et augmentation d'ATP en présence de 25 µM CAA). L'augmentation d'ATP
cellulaire en présence de 25 µM CAA, en hypoxie, ne peut pas être expliquée par
l'activation de la glycolyse induite par l'hypoxie. Une telle augmentation avait déjà été
observée en normoxie. Néanmoins, l'augmentation d'ATP en présence de 25 µM
CAA est moins importante en hypoxie qu'en normoxie (23% vs. 53% par rapport au
contrôle sans CAA, respectivement). Cela signifie que l'hypoxie, dans nos conditions,
inhibe seulement partiellement la phosphorylation oxydative.
Il a été déjà montré que l'hypoxie active l'HIF-1, un facteur de transcription qui
contrôle l'expression de la plupart des gènes impliqués dans la régulation d'hypoxie
et dans la croissance, le développement et les métastases des cancers [133, 134,
276]. Il stimule l'expression de pratiquement toutes les enzymes de la glycolyse ainsi
que le transporteur de glucose GLUT-1 [138]. Son élimination dans les cellules MCF-
7 réduit la croissance cellulaire [277]. Bien que l'HIF-1 n'ait pas été déterminé dans
cette étude, il est fort probable qu'il soit induit par l'hypoxie dans nos expériences.
Cette induction explique la surexpression des enzymes étudiées, et par conséquent,
l'augmentation de la consommation du glucose et de la production de lactate.
Il est probable que l'hypoxie de 5% O2, appliquée dans nos expériences, n'ait pas été
suffisamment sévère pour inhiber complètement la phosphorylation oxydative et, par
conséquent, abolir la production d'ATP dans les cellules MCF-7. L'effet du CAA sur
les cellules n'a pas pu être renforcé dans nos conditions. Néanmoins, l'hypoxie à 5%
O2 a été efficace, et cela s'est manifesté par l'induction des enzymes étudiées de la
glycolyse de manière suffisante pour activer cette voie métabolique.
D'autres expériences avec une hypoxie plus sévère (< 5% O2) sont envisagées afin
de voir si la combinaison de très faibles concentrations d'oxygène en présence de
CAA accroît l'efficacité de ce dernier sur l'inhibition de la prolifération des cellules
4. DES TRANCHES AUX CELLULES : COMPARAISON
Les deux parties de cette étude sont réalisées sur deux modèles expérimentaux
différents, les tranches de cortex rénal et les cellules de cancer du sein, MCF-7.
Nous pouvons, néanmoins, comparer ces deux modèles, ou plutôt comparer l'effet
du CAA sur les deux modèles.
Des concentrations faibles de CAA proches de celles trouvées chez les patients
traités par l'IFO s'avèrent toxiques sur les deux modèles. Les concentrations utilisées
sont comparables entre les deux modèles. La croissance des cellules MCF-7 est
inhibée en présence de 10 et 25 µM CAA qui est donc toxique sur les cellules
cancéreuses. La néphrotoxicité du CAA sur les tranches de cortex est montrée entre
25 et 75 µM CAA. Il convient de rappeler que des concentrations plasmatiques
comprises entre 5 et 200 µM CAA ont été déterminées lors de traitements par l'IFO.
Le CAA inhibe la voie métabolique principale du modèle étudié : la néoglucogenèse
dans les tranches de cortex rénal et la glycolyse dans les cellules MCF-7. L'inhibition
de ces deux voies métaboliques est accompagnée d'une inhibition de certaines
activités enzymatiques catalysant chacune des deux voies.
Un point commun de l'effet du CAA sur les deux modèles est l'inhibition de la
GAPDH, une enzyme commune entre la néoglucogenèse et la glycolyse. Elle est
inhibée de 53% dans les tranches de cortex rénal en présence de 75 µM CAA et de
62% dans les cellules MCF-7 incubées avec 25 µM CAA. C'est sur ce point commun
que nous nous sommes basés pour lier les deux parties de l'étude. Evidemment, les
résultats de la deuxième partie montrent que l'inhibition de la glycolyse dans les
cellules MCF-7 n'était probablement pas la conséquence directe de l'inhibition de la
GAPDH, mais plutôt celle du transport cellulaire du glucose et celle de l'HK.
Une différence importante entre les deux modèles concerne l'effet du CAA sur l'ATP
cellulaire. Dans les tranches de cortex, la concentration d'ATP est considérablement
diminuée en présence de 25 µM CAA après 24 heures d'exposition, tandis que dans
les cellules MCF-7, à la même concentration de CAA et au même temps d'exposition,
l'ATP cellulaire augmente de façon très importante (54%) malgré une inhibition de
45% de la glycolyse. Il est donc probable que la glycolyse ne soit pas la source
unique d'ATP dans les cellules MCF-7 produit également par voie oxydative. La
synthèse d'ATP par la phosphorylation oxydative semble beaucoup moins affectée
dans les cellules cancéreuses que dans les cellules rénales pour une même
concentration de CAA (25 µM). Dans ces dernières, une inhibition du complexe I de
la chaîne respiratoire semble être à l'origine de cette inhibition qui ne semble pas
être retrouvée dans les cellules MCF-7.
Les effets toxiques du CAA à la fois sur les cellules rénales et sur les cellules
cancéreuses MCF-7 portent à croire que le CAA n'est pas simplement responsable
de la néphrotoxicité de l'IFO, mais pourrait également contribuer, au moins en partie,
à l'efficacité thérapeutique de ce médicament.
VI. CONCLUSION GENERALE
Nous avons clairement montré dans cette étude que le CAA, utilisé à des
concentrations faibles (25 -75 µM) proches de celles rencontrées en pratique clinique,
exerce des effets toxiques sur le tissu rénal de rat. Cette néphrotoxicité s'est
manifestée par une chute du glutathion et des thiols cellulaires ainsi que par une
diminution de l'ATP. L'inhibition du métabolisme rénal du lactate, et par conséquent
l'inhibition de la néoglucogenèse à partir de ce substrat, sont également des aspects
de la néphrotoxicité du CAA. Nos résultats montrent que l'inhibition du complexe I de
la chaîne respiratoire mitochondriale est le responsable de l'inhibition de l'oxydation
de lactate sous l'effet du CAA. L'inhibition de la néoglucogenèse est liée à une
inhibition de certaines enzymes de cette voie métabolique notamment la GAPDH,
enzyme commune entre la néoglucogenèse et la glycolyse. Etant donné que la
glycolyse est la voie métabolique principale des cellules cancéreuses, et que
l'inhibition de cette voie est proposée par de nombreux auteurs pour inhiber la
prolifération de ces cellules, nous avons pensé que l'inhibition par le CAA de la
GAPDH, et ainsi de la glycolyse, pourrait avoir des conséquences sur la prolifération
des cellules cancéreuses.
Cette hypothèse s'est avérée incorrecte puisque l'inhibition induite par le CAA de la
GAPDH n'était pas responsable de l'inhibition de la glycolyse dans les cellules
cancéreuses utilisées dans cette étude (MCF-7). Néanmoins, nous avons montré
que le CAA (10 et 25 µM) inhibe la prolifération des cellules MCF-7, une inhibition
liée à l'inhibition de la glycolyse qui induit une diminution de la provision des
précurseurs des biosynthèses. L'inhibition de la glycolyse est expliquée par
l'inhibition du transport cellulaire du glucose ainsi que par l'inhibition de l'HK. D'une
manière surprenante, l'inhibition de la prolifération cellulaire n'était pas accompagnée
d'une chute de l'ATP. Au contraire, ce dernier a augmenté en présence de 25 µM
CAA, la concentration qui inhibe complètement la prolifération cellulaire.
L'augmentation de l'ATP est donc expliquée par l'absence de son utilisation pour la
prolifération cellulaire. Si l'inhibition de la glycolyse n'a pas induit une chute de l'ATP,
ce dernier pourrait donc avoir d'autres sources, notamment l'oxydative.
Le CAA est donc bien impliqué dans le mécanisme de la néphrotoxicité de l'IFO,
mais il pourrait également contribuer à son effet thérapeutique.
REFERENCES BIBLIOGRAPHIQUES
Paillard M. Rein. In: Physiologie Humaine, 2ème edn. Guénard H, editors. Editions Pradel, Paris (1996). vol I, pp 241-58.
Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care (2001) 24:382-91.
Curthoys NP, Lowry OH. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem (1973) 248:162-8.
Mandel LJ. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu Rev Physiol (1985) 47:85-101.
Stryer L. Chapitre 18: La voie de pentose phosphate et la néoglucogenèse. In: La Biochimie de Lubert Stryer, 3éme edn. Flammarion, Paris (1992). pp 427-48.
Stryer L. Chapitre 15: La glycolyse. In: La Biochimie de Lubert Stryer, 3éme edn. Flammarion, Paris (1992). pp 350-72.
Gilman A. The initial clinical trial of nitrogen mustard. Am J Surg (1963) 105:574-8.
Clavel B, Mathe G, Hayat M. [Phase II therapeutic trial (screening) of ifosfamide in hematosarcomas and solid tumors]. Sem Hop Ther (1975) 51:7-10.
Cohen MH, Creaven PJ, Tejada F, Hansen HH, Muggia F, et al. Phase I clinical trial of isophosphamide (NSC-109724). Cancer Chemother Rep (1975) 59:751-5.
Czownicki Z, Utracka B, Till A, Styrna M. Clinical evaluation of ifosfamide in the treatment of malignant neoplasms. Nowotwory (1973) 23:285-90.
Furlanut M, Franceschi L. Pharmacology of ifosfamide. Oncology (2003) 65 Suppl 2:2-6.
Lokiec F. Ifosfamide: pharmacokinetic properties for central nervous system metastasis prevention. Ann Oncol (2006) 17 Suppl 4:iv33-6.
Zhang J, Tian Q, Yung Chan S, Chuen Li S, Zhou S, et al. Metabolism and transport of oxazaphosphorines and the clinical implications. Drug Metab Rev (2005) 37:611-703.
Carli M, Passone E, Perilongo G, Bisogno G. Ifosfamide in pediatric solid tumors. Oncology (2003) 65 Suppl 2:99-104.
Hartmann JT, Gauler T, Metzner B, Gerl A, Casper J, et al. Phase I/II study of sequential dose-intensified ifosfamide, cisplatin, and etoposide plus paclitaxel as induction chemotherapy for poor prognosis germ cell tumors by the German Testicular Cancer Study Group. J Clin Oncol (2007) 25:5742-7.
Marangolo M, Giovanis P. Ifosfamide in small cell lung cancer. Oncology (2003) 65 Suppl 2:46-9.
Park S, Ahn MJ, Ahn JS, Lee J, Hong YS, et al. Combination chemotherapy with paclitaxel and ifosfamide as the third-line regimen in patients with heavily pretreated small cell lung cancer. Lung Cancer (2007) 58:116-22.
Piantedosi FV, Caputo F, Mazzarella G, Gilli M, Pontillo A, et al. Gemcitabine, ifosfamide and paclitaxel in advanced/metastatic non-small cell lung cancer patients: a phase II study. Cancer Chemother Pharmacol (2008) 61:803-7.
Schutt P, Passon J, Ebeling P, Welt A, Muller S, et al. Ifosfamide, etoposide, cytarabine, and dexamethasone as salvage treatment followed by high-dose cyclophosphamide, melphalan, and etoposide with autologous peripheral blood stem cell transplantation for relapsed or refractory lymphomas. Eur J Haematol (2007) 78:93-101.
Tascilar M, Loos WJ, Seynaeve C, Verweij J, Sleijfer S. The pharmacologic basis of ifosfamide use in adult patients with advanced soft tissue sarcomas. Oncologist (2007) 12:1351-60.
Verma S, Younus J, Stys-Norman D, Haynes AE, Blackstein M. Ifosfamide-based combination chemotherapy in advanced soft-tissue sarcoma: a practice guideline. Curr Oncol (2007) 14:144-8.
Kosmas C, Tsavaris N, Mylonakis N, Tsakonas G, Gassiamis A, et al. Docetaxel-ifosfamide combination in patients with advanced breast cancer failing prior anthracycline-based regimens: results of a phase I-II study. J Chemother (2007) 19:322-31.
Sutton G, Kauderer J, Carson LF, Lentz SS, Whitney CW, et al. Adjuvant ifosfamide and cisplatin in patients with completely resected stage I or II carcinosarcomas (mixed mesodermal tumors) of the uterus: a Gynecologic Oncology Group study. Gynecol Oncol (2005) 96:630-4.
Cicin I, Saip P, Eralp Y, Selam M, Topuz S, et al. Ovarian carcinosarcomas: clinicopathological prognostic factors and evaluation of chemotherapy regimens containing platinum. Gynecol Oncol (2008) 108:136-40.
Crotzer DR, Wolf JK, Gano JB, Gershenson DM, Levenback C. A pilot study of cisplatin, ifosfamide and mesna in the treatment of malignant mixed mesodermal tumors of the ovary. Gynecol Oncol (2007) 105:399-403.
Basaran M, Bavbek ES, Saglam S, Eralp L, Sakar B, et al. A phase II study of cisplatin, ifosfamide and epirubicin combination chemotherapy in adults with nonmetastatic and extremity osteosarcomas. Oncology (2007) 72:255-60.
Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med (2003) 348:694-701.
Jurgens H, Treuner J, Winkler K, Gobel U. Ifosfamide in pediatric malignancies. Semin Oncol (1989) 16:46-50.
Voute PA, van den Berg H, Behrendt H, Michiels E, de Kraker J. Ifosfamide in the treatment of pediatric malignancies. Semin Oncol (1996) 23:8-11.
Brain EG, Rezai K, Weill S, Gauzan MF, Santoni J, et al. Variations in schedules of ifosfamide administration: a better understanding of its implications on pharmacokinetics through a randomized cross-over study. Cancer Chemother Pharmacol (2007) 60:375-81.
Lewis LD. Ifosfamide pharmacokinetics. Invest New Drugs (1991) 9:305-11.
Lewis LD, Fitzgerald DL, Mohan P, Thatcher N, Harper PG, et al. The pharmacokinetics of ifosfamide given as short and long intravenous infusions in cancer patients. Br J Clin Pharmacol (1991) 31:77-82.
Pendyala L, Creaven PJ, Schwartz G, Meropol NJ, Bolanowska-Higdon W, et al. Intravenous ifosfamide/mesna is associated with depletion of plasma thiols without depletion of leukocyte glutathione. Clin Cancer Res (2000) 6:1314-21.
Klastersky J. Side effects of ifosfamide. Oncology (2003) 65 Suppl 2:7-10.
Lima MV, Ferreira FV, Macedo FY, de Castro Brito GA, Ribeiro RA. Histological changes in bladders of patients submitted to ifosfamide chemotherapy even with mesna prophylaxis. Cancer Chemother Pharmacol (2007) 59:643-50.
Dechant KL, Brogden RN, Pilkington T, Faulds D. Ifosfamide/mesna. A review of its antineoplastic activity, pharmacokinetic properties and therapeutic efficacy in cancer. Drugs (1991) 42:428-67.
Andriole GL, Sandlund JT, Miser JS, Arasi V, Linehan M, et al. The efficacy of mesna (2-mercaptoethane sodium sulfonate) as a uroprotectant in patients with hemorrhagic cystitis receiving further oxazaphosphorine chemotherapy. J Clin Oncol (1987) 5:799-803.
Brock N, Pohl J, Stekar J. Detoxification of urotoxic oxazaphosphorines by sulfhydryl compounds. J Cancer Res Clin Oncol (1981) 100:311-20.
Batista CK, Mota JM, Souza ML, Leitao BT, Souza MH, et al. Amifostine and glutathione prevent ifosfamide- and acrolein-induced hemorrhagic cystitis. Cancer Chemother Pharmacol (2007) 59:71-7.
Mota JM, Brito GA, Loiola RT, Cunha FQ, Ribeiro Rde A. Interleukin-11 attenuates ifosfamide-induced hemorrhagic cystitis. Int Braz J Urol (2007) 33:704-10.
Vieira MM, Macedo FY, Filho JN, Costa AC, Cunha AN, et al. Ternatin, a flavonoid, prevents cyclophosphamide and ifosfamide-induced hemorrhagic cystitis in rats. Phytother Res (2004) 18:135-41.
Aleksa K, Halachmi N, Ito S, Koren G. A tubule cell model for ifosfamide nephrotoxicity. Can J Physiol Pharmacol (2005) 83:499-508.
Nissim I, Horyn O, Daikhin Y, Luhovyy B, Phillips PC, et al. Ifosfamide-induced nephrotoxicity: mechanism and prevention. Cancer Res (2006) 66:7824-31.
Skinner R. Chronic ifosfamide nephrotoxicity in children. Med Pediatr Oncol (2003) 41:190-7.
Skinner R, Sharkey IM, Pearson AD, Craft AW. Ifosfamide, mesna, and nephrotoxicity in children. J Clin Oncol (1993) 11:173-90.
Loebstein R, Atanackovic G, Bishai R, Wolpin J, Khattak S, et al. Risk factors for long-term outcome of ifosfamide-induced nephrotoxicity in children. J Clin Pharmacol (1999) 39:454-61.
Marina NM, Poquette CA, Cain AM, Jones D, Pratt CB, et al. Comparative renal tubular toxicity of chemotherapy regimens including ifosfamide in patients with newly diagnosed sarcomas. J Pediatr Hematol Oncol (2000) 22:112-8.
Patterson WP, Khojasteh A. Ifosfamide-induced renal tubular defects. Cancer (1989) 63:649-51.
Skinner R, Pearson AD, English MW, Price L, Wyllie RA, et al. Risk factors for ifosfamide nephrotoxicity in children. Lancet (1996) 348:578-80.
Ashraf MS, Brady J, Breatnach F, Deasy PF, O'Meara A. Ifosfamide nephrotoxicity in paediatric cancer patients. Eur J Pediatr (1994) 153:90-4.
Lee BS, Lee JH, Kang HG, Hahn H, Shin HY, et al. Ifosfamide nephrotoxicity in pediatric cancer patients. Pediatr Nephrol (2001) 16:796-9.
Stohr W, Paulides M, Bielack S, Jurgens H, Treuner J, et al. Ifosfamide-induced nephrotoxicity in 593 sarcoma patients: a report from the Late Effects Surveillance System. Pediatr Blood Cancer (2007) 48:447-52.
Rossi R, Godde A, Kleinebrand A, Riepenhausen M, Boos J, et al. Unilateral nephrectomy and cisplatin as risk factors of ifosfamide-induced nephrotoxicity: analysis of 120 patients. J Clin Oncol (1994) 12:159-65.
Skinner R, Cotterill SJ, Stevens MC. Risk factors for nephrotoxicity after ifosfamide treatment in children: a UKCCSG Late Effects Group study. United Kingdom Children's Cancer Study Group. Br J Cancer (2000) 82:1636-45.
Skinner R, Pearson AD, Price L, Coulthard MG, Craft AW. Nephrotoxicity after ifosfamide. Arch Dis Child (1990) 65:732-8.
Rossi R. Nephrotoxicity of ifosfamide-moving towards understanding the molecular mechanisms. Nephrol Dial Transplant (1997) 12:1091-2.
Rossi R, Pleyer J, Schafers P, Kuhn N, Kleta R, et al. Development of ifosfamide-induced nephrotoxicity: prospective follow-up in 75 patients. Med Pediatr Oncol (1999) 32:177-82.
De Schepper J, Stevens G, Verboven M, Baeta C, Otten J. Ifosfamide-induced Fanconi's syndrome with growth failure in a 2-year-old child. Am J Pediatr Hematol Oncol (1991) 13:39-41.
Stohr W, Patzer L, Paulides M, Kremers A, Beck JD, et al. Growth impairment after ifosfamide-induced nephrotoxicity in children. Pediatr Blood Cancer (2007) 48:571-6.
Rossi R. Nephrotoxicity of cytostatic therapy. Nephrol Dial Transplant (1996) 11:1092-6.
Rossi R, Kleta R, Ehrich JH. Renal involvement in children with malignancies. Pediatr Nephrol (1999) 13:153-62.
Loebstein R, Koren G. Ifosfamide-induced nephrotoxicity in children: critical review of predictive risk factors. Pediatrics (1998) 101:E8.
Rossi R, Danzebrink S, Hillebrand D, Linnenburger K, Ullrich K, et al. Ifosfamide-induced subclinical nephrotoxicity and its potentiation by cisplatinum. Med Pediatr Oncol (1994) 22:27-32.
Aleksa K, Halachmi N, Ito S, Koren G. Renal ontogeny of ifosfamide nephrotoxicity. J Lab Clin Med (2004) 144:285-93.
McCune JS, Friedman DL, Schuetze S, Blough D, Magbulos M, et al. Influence of age upon Ifosfamide-induced nephrotoxicity. Pediatr Blood Cancer (2004) 42:427-32.
Goren MP, Wright RK, Horowitz ME, Pratt CB. Ifosfamide-induced subclinical tubular nephrotoxicity despite mesna. Cancer Treat Rep (1987) 71:127-30.
Mohrmann M, Ansorge S, Schonfeld B, Brandis M. Dithio-bis-mercaptoethanesulphonate (DIMESNA) does not prevent cellular damage by metabolites of ifosfamide and cyclophosphamide in LLC-PK1 cells. Pediatr Nephrol (1994) 8:458-65.
Sangster G, Kaye SB, Calman KC, Dalton JF. Failure of 2-mercaptoethane sulphonate sodium (mesna) to protect against ifosfamide nephrotoxicity. Eur J Cancer Clin Oncol (1984) 20:435-6.
Yaseen Z, Michoudet C, Baverel G, Dubourg L. In vivo mesna and amifostine do not prevent chloroacetaldehyde nephrotoxicity in vitro. Pediatr Nephrol (2008) 23:611-8.
Hartmann JT, Knop S, Fels LM, van Vangerow A, Stolte H, et al. The use of reduced doses of amifostine to ameliorate nephrotoxicity of cisplatin/ifosfamide-based chemotherapy in patients with solid tumors. Anticancer Drugs (2000) 11:1-6.
de Kraker J, Bierings MB, Offringa M. Lack of protection of proximal tubular cells by amifostine (ethyol) in ifosfamide-containing regimens. Med Pediatr Oncol (2000) 34:78-9.
Petrilli AS, Oliveira DT, Ginani VC, Kechichian R, Dishtchekenian A, et al. Use of amifostine in the therapy of osteosarcoma in children and adolescents. J Pediatr Hematol Oncol (2002) 24:188-91.
Zaki EL, Springate JE, Taub M. Comparative toxicity of ifosfamide metabolites and protective effect of mesna and amifostine in cultured renal tubule cells. Toxicol In Vitro (2003) 17:397-402.
Chen N, Aleksa K, Woodland C, Rieder M, Koren G. N-Acetylcysteine prevents ifosfamide-induced nephrotoxicity in rats. Br J Pharmacol (2008) 153:1364-72.
Chen N, Aleksa K, Woodland C, Rieder M, Koren G. The effect of N-acetylcysteine on ifosfamide-induced nephrotoxicity: in vitro studies in renal tubular cells. Transl Res (2007) 150:51-7.
Schwerdt G, Gordjani N, Benesic A, Freudinger R, Wollny B, et al. Chloroacetaldehyde- and acrolein-induced death of human proximal tubule cells. Pediatr Nephrol (2006) 21:60-7.
Springate J, Chan K, Lu H, Davies S, Taub M. Toxicity of ifosfamide and its metabolite chloroacetaldehyde in cultured renal tubule cells. In Vitro Cell Dev Biol Anim (1999) 35:314-7.
Zamlauski-Tucker MJ, Morris ME, Springate JE. Ifosfamide metabolite chloroacetaldehyde causes Fanconi syndrome in the perfused rat kidney. Toxicol Appl Pharmacol (1994) 129:170-5.
Benesic A, Schwerdt G, Freudinger R, Mildenberger S, Groezinger F, et al. Chloroacetaldehyde as a sulfhydryl reagent: the role of critical thiol groups in ifosfamide nephropathy. Kidney Blood Press Res (2006) 29:280-93.
Springate JE. Ifosfamide metabolite chloroacetaldehyde causes renal dysfunction in vivo. J Appl Toxicol (1997) 17:75-9.
Kaijser GP, De Kraker J, Bult A, Underberg WJ, Beijnen JH. Pharmacokinetics of ifosfamide and some metabolites in children. Anticancer Res (1998) 18:1941-9.
Kupfer A, Aeschlimann C, Wermuth B, Cerny T. Prophylaxis and reversal of ifosfamide encephalopathy with methylene-blue. Lancet (1994) 343:763-4.
Park IS, Lee HJ, Lee YS, Hwang JS, Lee MS. Ifosfamide-induced encephalopathy with or without using methylene blue. Int J Gynecol Cancer (2005) 15:807-10.
Aeschlimann C, Cerny T, Kupfer A. Inhibition of (mono)amine oxidase activity and prevention of ifosfamide encephalopathy by methylene blue. Drug Metab Dispos (1996) 24:1336-9.
Pelgrims J, De Vos F, Van den Brande J, Schrijvers D, Prove A, et al. Methylene blue in the treatment and prevention of ifosfamide-induced encephalopathy: report of 12 cases and a review of the literature. Br J Cancer (2000) 82:291-4.
Turner AR, Duong CD, Good DJ. Methylene blue for the treatment and prophylaxis of ifosfamide-induced encephalopathy. Clin Oncol (R Coll Radiol) (2003) 15:435-9.
Hamadani M, Awan F. Role of thiamine in managing ifosfamide-induced encephalopathy. J Oncol Pharm Pract (2006) 12:237-9.
Allen A. The cardiotoxicity of chemotherapeutic drugs. Semin Oncol (1992) 19:529-42.
Aleksa K, Matsell D, Krausz K, Gelboin H, Ito S, et al. Cytochrome P450 3A and 2B6 in the developing kidney: implications for ifosfamide nephrotoxicity. Pediatr Nephrol (2005) 20:872-85.
Springate J, Zamlauski-Tucker MJ, Lu H, Chan KK. Renal clearance of ifosfamide. Drug Metab Dispos (1997) 25:1081-2.
Woodland C, Ito S, Granvil CP, Wainer IW, Klein J, et al. Evidence of renal metabolism of ifosfamide to nephrotoxic metabolites. Life Sci (2000) 68:109-17.
Granvil CP, Madan A, Sharkawi M, Parkinson A, Wainer IW. Role of CYP2B6 and CYP3A4 in the in vitro N-dechloroethylation of (R)- and (S)-ifosfamide in human liver microsomes. Drug Metab Dispos (1999) 27:533-41.
Boddy AV, Yule SM. Metabolism and pharmacokinetics of oxazaphosphorines. Clin Pharmacokinet (2000) 38:291-304.
Colvin M. The comparative pharmacology of cyclophosphamide and ifosfamide. Semin Oncol (1982) 9:2-7.
Kaijser GP, Korst A, Beijnen JH, Bult A, Underberg WJ. The analysis of ifosfamide and its metabolites (review). Anticancer Res (1993) 13:1311-24.
Wagner T. Ifosfamide clinical pharmacokinetics. Clin Pharmacokinet (1994) 26:439-56.
Sarosy G. Ifosfamide--pharmacologic overview. Semin Oncol (1989) 16:2-8.
Bramwell VH, Mouridsen HT, Santoro A, Blackledge G, Somers R, et al. Cyclophosphamide versus ifosfamide: a randomized phase II trial in adult soft-tissue sarcomas. The European Organization for Research and Treatment of Cancer [EORTC], Soft Tissue and Bone Sarcoma Group. Cancer Chemother Pharmacol (1993) 31 Suppl 2:S180-4.
Brade W, Seeber S, Herdrich K. Comparative activity of ifosfamide and cyclophosphamide. Cancer Chemother Pharmacol (1986) 18 Suppl 2:S1-9.
[100] Bramwell VH, Mouridsen HT, Santoro A, Blackledge G, Somers R, et al.
Cyclophosphamide versus ifosfamide: preliminary report of a randomized phase II trial in adult soft tissue sarcomas. Cancer Chemother Pharmacol (1986) 18 Suppl 2:S13-6.
[101] Bramwell VH, Mouridsen HT, Santoro A, Blackledge G, Somers R, et al.
Cyclophosphamide versus ifosfamide: final report of a randomized phase II trial in adult soft tissue sarcomas. Eur J Cancer Clin Oncol (1987) 23:311-21.
[102] Chen CS, Jounaidi Y, Waxman DJ. Enantioselective metabolism and cytotoxicity of
R-ifosfamide and S-ifosfamide by tumor cell-expressed cytochromes P450. Drug Metab Dispos (2005) 33:1261-7.
[103] Roy P, Tretyakov O, Wright J, Waxman DJ. Stereoselective metabolism of ifosfamide
by human P-450s 3A4 and 2B6. Favorable metabolic properties of R-enantiomer. Drug Metab Dispos (1999) 27:1309-18.
[104] Lu H, Wang JJ, Chan KK, Philip PA. Stereoselectivity in metabolism of ifosfamide by
CYP3A4 and CYP2B6. Xenobiotica (2006) 36:367-85.
[105] Wang JJ, Lu H, Chan KK. Stereoselective pharmacokinetics of ifosfamide in male
and female rats. AAPS PharmSci (2000) 2:E17.
[106] Aleksa K, Ito S, Koren G. Renal-tubule metabolism of ifosfamide to the nephrotoxic
chloroacetaldehyde: pharmacokinetic modeling for estimation of intracellular levels. J Lab Clin Med (2004) 143:159-62.
[107] Aleksa K, Ito S, Koren G. Enantioselective metabolism of ifosfamide by the kidney.
Chirality (2006) 18:398-405.
[108] Bruggemann SK, Kisro J, Wagner T. Ifosfamide cytotoxicity on human tumor and
renal cells: role of chloroacetaldehyde in comparison to 4-hydroxyifosfamide. Cancer Res (1997) 57:2676-80.
[109] Goren MP, Wright RK, Pratt CB, Pell FE. Dechloroethylation of ifosfamide and
neurotoxicity. Lancet (1986) 2:1219-20.
[110] Kaijser GP, Beijnen JH, Jeunink EL, Bult A, Keizer HJ, et al. Determination of
chloroacetaldehyde, a metabolite of oxazaphosphorine cytostatic drugs, in plasma. J Chromatogr (1993) 614:253-9.
[111] Kurowski V, Cerny T, Kupfer A, Wagner T. Metabolism and pharmacokinetics of oral
and intravenous ifosfamide. J Cancer Res Clin Oncol (1991) 117 Suppl 4:S148-53.
[112] Mohrmann M, Ansorge S, Schmich U, Schonfeld B, Brandis M. Toxicity of ifosfamide,
cyclophosphamide and their metabolites in renal tubular cells in culture. Pediatr Nephrol (1994) 8:157-63.
[113] Dubourg L, Michoudet C, Cochat P, Baverel G. Human kidney tubules detoxify
chloroacetaldehyde, a presumed nephrotoxic metabolite of ifosfamide. J Am Soc Nephrol (2001) 12:1615-23.
[114] Springate J, Taub M. Ifosfamide toxicity in cultured proximal renal tubule cells.
Pediatr Nephrol (2007) 22:358-65.
[115] Dubourg L, Taniere P, Cochat P, Baverel G, Michoudet C. Toxicity of
chloroacetaldehyde is similar in adult and pediatric kidney tubules. Pediatr Nephrol (2002) 17:97-103.
[116] Lind MJ, McGown AT, Hadfield JA, Thatcher N, Crowther D, et al. The effect of
ifosfamide and its metabolites on intracellular glutathione levels in vitro and in vivo. Biochem Pharmacol (1989) 38:1835-40.
[117] Benesic A, Schwerdt G, Mildenberger S, Freudinger R, Gordjani N, et al. Disturbed
Ca2+-signaling by chloroacetaldehyde: a possible cause for chronic ifosfamide nephrotoxicity. Kidney Int (2005) 68:2029-41.
[118] Patzer L, Hernando N, Ziegler U, Beck-Schimmer B, Biber J, et al. Ifosfamide
metabolites CAA, 4-OH-Ifo and Ifo-mustard reduce apical phosphate transport by changing NaPi-IIa in OK cells. Kidney Int (2006) 70:1725-34.
[119] Mohrmann M, Kupper N, Schonfield B, Brandis M. Ifosfamide and mesna: effects on
the Na/H exchanger activity in renal epithelial cells in culture (LLC-PK1). Ren Physiol Biochem (1995) 18:118-27.
[120] Mohrmann M, Pauli A, Ritzer M, Schonfeld B, Seifert B, et al. Inhibition of sodium-
dependent transport systems in LLC-PK1 cells by metabolites of ifosfamide. Ren Physiol Biochem (1992) 15:289-301.
[121] Sood C, O'Brien PJ. Molecular mechanisms of chloroacetaldehyde-induced
cytotoxicity in isolated rat hepatocytes. Biochem Pharmacol (1993) 46:1621-6.
[122] Racker E. History of the Pasteur effect and its pathobiology. Mol Cell Biochem (1974)
[123] Warburg O. On the origin of cancer cells. Science (1956) 123:309-14. [124] Gatenby RA. The potential role of transformation-induced metabolic changes in
tumor-host interaction. Cancer Res (1995) 55:4151-6.
[125] Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev
Cancer (2004) 4:891-9.
[126] Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol (2006) 18:598-608. [127] Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys
Res Commun (2004) 313:459-65.
[128] Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer
therapeutic implications. J Bioenerg Biomembr (2007) 39:267-74.
[129] Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment.
Oncogene (2006) 25:4633-46.
[130] Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene
(2006) 25:4647-62.
[131] Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp
Quant Biol (2005) 70:363-74.
[132] Rodriguez-Enriquez S, Torres-Marquez ME, Moreno-Sanchez R. Substrate oxidation
and ATP supply in AS-30D hepatoma cells. Arch Biochem Biophys (2000) 375:21-30.
[133] Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg's effect. J
Bioenerg Biomembr (2007) 39:223-9.
[134] Maxwell PH, Pugh CW, Ratcliffe PJ. Activation of the HIF pathway in cancer. Curr
Opin Genet Dev (2001) 11:293-9.
[135] Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the
HIF hydroxylase pathway. Mol Cell (2008) 30:393-402.
[136] Brahimi-Horn MC, Pouyssegur J. The hypoxia-inducible factor and tumor progression
along the angiogenic pathway. Int Rev Cytol (2005) 242:157-213.
[137] Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, et al. Up-
regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol (2005) 206:291-304.
[138] Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes
encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem (1994) 269:23757-63.
[139] Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of
pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177-85.
[140] Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation
to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187-97.
[141] Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell (2008)
[142] Revillion F, Pawlowski V, Hornez L, Peyrat JP. Glyceraldehyde-3-phosphate
dehydrogenase gene expression in human breast cancer. Eur J Cancer (2000) 36:1038-42.
[143] Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular
events underlying one of cancers' most common phenotypes, the "Warburg Effect", i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr (2007) 39:211-22.
[144] Newsholme EA, Board M. Application of metabolic-control logic to fuel utilization and
its significance in tumor cells. Adv Enzyme Regul (1991) 31:225-46.
[145] Mazurek S, Boschek CB, Eigenbrodt E. The role of phosphometabolites in cell
proliferation, energy metabolism, and tumor therapy. J Bioenerg Biomembr (1997) 29:315-30.
[146] Mazurek S, Hugo F, Failing K, Eigenbrodt E. Studies on associations of glycolytic and
glutaminolytic enzymes in MCF-7 cells: role of P36. J Cell Physiol (1996) 167:238-50.
[147] Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect:
the metabolic requirements of cell proliferation. Science (2009) 324:1029-33.
[148] Maublant J, Vuillez JP, Talbot JN, Lumbroso J, Muratet JP, et al. [Positron emission
tomography (PET) and (F-18)-fluorodeoxyglucose in (FDG) in cancerology]. Bull Cancer (1998) 85:935-50.
[149] Board M, Humm S, Newsholme EA. Maximum activities of key enzymes of glycolysis,
glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J (1990) 265:503-9.
[150] Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E. Pyruvate kinase type
M2: a crossroad in the tumor metabolome. Br J Nutr (2002) 87 Suppl 1:S23-9.
[151] Linden M, Gellerfors P, Nelson BD. Pore protein and the hexokinase-binding protein
from the outer membrane of rat liver mitochondria are identical. FEBS Lett (1982) 141:189-92.
[152] Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells. Isolation,
sequence, and activity of the promoter for type II hexokinase. J Biol Chem (1995) 270:16918-25.
[153] Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells.
Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem (1981) 256:8699-704.
[154] Katabi MM, Chan HL, Karp SE, Batist G. Hexokinase type II: a novel tumor-specific
promoter for gene-targeted therapy differentially expressed and regulated in human cancer cells. Hum Gene Ther (1999) 10:155-64.
[155] Marin-Hernandez A, Rodriguez-Enriquez S, Vital-Gonzalez PA, Flores-Rodriguez FL,
Macias-Silva M, et al. Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J (2006) 273:1975-88.
[156] Brown RS, Goodman TM, Zasadny KR, Greenson JK, Wahl RL. Expression of
hexokinase II and Glut-1 in untreated human breast cancer. Nucl Med Biol (2002) 29:443-53.
[157] Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism
in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res (1996) 56:2468-71.
[158] Vora S, Halper JP, Knowles DM. Alterations in the activity and isozymic profile of
human phosphofructokinase during malignant transformation in vivo and in vitro: transformation- and progression-linked discriminants of malignancy. Cancer Res (1985) 45:2993-3001.
[159] Ibsen KH, Orlando RA, Garratt KN, Hernandez AM, Giorlando S, et al. Expression of
multimolecular forms of pyruvate kinase in normal, benign, and malignant human breast tissue. Cancer Res (1982) 42:888-92.
[160] Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, et al.
The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature (2008) 452:230-U74.
[161] Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate
kinase M2 allosteric regulation and catalysis. Biochemistry (2005) 44:9417-29.
[162] Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role
in tumor growth and spreading. Semin Cancer Biol (2005) 15:300-8.
[163] Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2
is a phosphotyrosine-binding protein. Nature (2008) 452:181-6.
[164] Nelson JA, Falk RE. The efficacy of phloridzin and phloretin on tumor cell growth.
Anticancer Res (1993) 13:2287-92.
[165] Waki A, Kato H, Yano R, Sadato N, Yokoyama A, et al. The importance of glucose
transport activity as the rate-limiting step of 2-deoxyglucose uptake in tumor cells in vitro. Nucl Med Biol (1998) 25:593-7.
[166] Brown RS, Wahl RL. Overexpression of Glut-1 glucose transporter in human breast
cancer. An immunohistochemical study. Cancer (1993) 72:2979-85.
[167] Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose
transporter (GLUT) proteins in cancer. J Cell Physiol (2005) 202:654-62.
[168] Younes M, Brown RW, Mody DR, Fernandez L, Laucirica R. GLUT1 expression in
human breast carcinoma: correlation with known prognostic markers. Anticancer Res (1995) 15:2895-8.
[169] Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Wide expression of
the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res (1996) 56:1164-7.
[170] Grover-McKay M, Walsh SA, Seftor EA, Thomas PA, Hendrix MJ. Role for glucose
transporter 1 protein in human breast cancer. Pathol Oncol Res (1998) 4:115-20.
[171] Rogers S, Macheda ML, Docherty SE, Carty MD, Henderson MA, et al. Identification
of a novel glucose transporter-like protein-GLUT-12. Am J Physiol Endocrinol Metab (2002) 282:E733-8.
[172] Rogers S, Docherty SE, Slavin JL, Henderson MA, Best JD. Differential expression of
GLUT12 in breast cancer and normal breast tissue. Cancer Lett (2003) 193:225-33.
[173] Birnbaum MJ, Haspel HC, Rosen OM. Transformation of rat fibroblasts by FSV
rapidly increases glucose transporter gene transcription. Science (1987) 235:1495-8.
[174] Burgman P, Odonoghue JA, Humm JL, Ling CC. Hypoxia-Induced increase in FDG
uptake in MCF7 cells. J Nucl Med (2001) 42:170-5.
[175] Clavo AC, Brown RS, Wahl RL. Fluorodeoxyglucose uptake in human cancer cell
lines is increased by hypoxia. J Nucl Med (1995) 36:1625-32.
[176] Birrell SN, Bentel JM, Hickey TE, Ricciardelli C, Weger MA, et al. Androgens induce
divergent proliferative responses in human breast cancer cell lines. J Steroid Biochem Mol Biol (1995) 52:459-67.
[177] Rivenzon-Segal D, Boldin-Adamsky S, Seger D, Seger R, Degani H. Glycolysis and
glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int J Cancer (2003) 107:177-82.
[178] Merrall NW, Plevin R, Gould GW. Growth factors, mitogens, oncogenes and the
regulation of glucose transport. Cell Signal (1993) 5:667-75.
[179] Wilding G, Lippman ME, Gelmann EP. Effects of steroid hormones and peptide
growth factors on protooncogene c-fos expression in human breast cancer cells. Cancer Res (1988) 48:802-5.
[180] Liu H, Hu YP, Savaraj N, Priebe W, Lampidis TJ. Hypersensitization of tumor cells to
glycolytic inhibitors. Biochemistry (2001) 40:5542-7.
[181] Munoz-Pinedo C, Ruiz-Ruiz C, Ruiz de Almodovar C, Palacios C, Lopez-Rivas A.
Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J Biol Chem (2003) 278:12759-68.
[182] Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, et al. Nitroxyl inhibits breast tumor
growth and angiogenesis. Int J Cancer (2008) 122:1905-10.
[183] Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme
inhibitors in cancer treatment. Expert Opin Investig Drugs (2008) 17:1533-45.
[184] Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721-32. [185] Spitz GA, Furtado CM, Sola-Penna M, Zancan P. Acetylsalicylic acid and salicylic
acid decrease tumor cell viability and glucose metabolism modulating 6-phosphofructo-1-kinase structure and activity. Biochem Pharmacol (2009) 77:46-53.
[186] Du GJ, Song ZH, Lin HH, Han XF, Zhang S, et al. Luteolin as a glycolysis inhibitor
offers superior efficacy and lesser toxicity of doxorubicin in breast cancer cells. Biochem Biophys Res Commun (2008) 372:497-502.
[187] Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, et al. 2-deoxy-D-
glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res (2004) 64:31-4.
[188] Maher JC, Krishan A, Lampidis TJ. Greater cell cycle inhibition and cytotoxicity
induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol (2004) 53:116-22.
[189] Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL. Novel therapy for
liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res (2002) 62:3909-13.
[190] Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, et al. Advanced cancers:
eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun (2004) 324:269-75.
[191] Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, et al. Synergistic effect of
targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia (2005) 19:2153-8.
[192] Buijs M, Vossen JA, Geschwind JF, Ishimori T, Engles JM, et al. Specificity of the
anti-glycolytic activity of 3-bromopyruvate confirmed by FDG uptake in a rat model of breast cancer. Invest New Drugs (2009) 27:120-3.
[193] Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, et al. Lonidamine, a
selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst (1981) 66:497-9.
[194] Rosbe KW, Brann TW, Holden SA, Teicher BA, Frei E, 3rd. Effect of lonidamine on
the cytotoxicity of four alkylating agents in vitro. Cancer Chemother Pharmacol (1989) 25:32-6.
[195] Chesney J. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell
glycolysis. Curr Opin Clin Nutr Metab Care (2006) 9:535-9.
[196] Song S, Finkel T. GAPDH and the search for alternative energy. Nat Cell Biol (2007)
[197] Sirover MA. Role of the glycolytic protein, glyceraldehyde-3-phosphate
dehydrogenase, in normal cell function and in cell pathology. J Cell Biochem (1997) 66:133-40.
[198] Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-
phosphate dehydrogenase, in mammalian cells. J Cell Biochem (2005) 95:45-52.
[199] Langbein S, Zerilli M, Zur Hausen A, Staiger W, Rensch-Boschert K, et al. Expression
of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br J Cancer (2006) 94:578-85.
[200] Rais B, Comin B, Puigjaner J, Brandes JL, Creppy E, et al. Oxythiamine and
dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich's tumor cells through inhibition of the pentose cycle. FEBS Lett (1999) 456:113-8.
[201] Penkowa M, Quintana A, Carrasco J, Giralt M, Molinero A, et al. Metallothionein
prevents neurodegeneration and central nervous system cell death after treatment with gliotoxin 6-aminonicotinamide. J Neurosci Res (2004) 77:35-53.
[202] Serkova N, Boros LG. Detection of resistance to imatinib by metabolic profiling:
clinical and drug development implications. Am J Pharmacogenomics (2005) 5:293-302.
[203] Briasoulis E, Pavlidis N, Terret C, Bauer J, Fiedler W, et al. Glufosfamide
administered using a 1-hour infusion given as first-line treatment for advanced pancreatic cancer. A phase II trial of the EORTC-new drug development group. Eur J Cancer (2003) 39:2334-40.
[204] Giaccone G, Smit EF, de Jonge M, Dansin E, Briasoulis E, et al. Glufosfamide
administered by 1-hour infusion as a second-line treatment for advanced non-small cell lung cancer; a phase II trial of the EORTC-New Drug Development Group. Eur J Cancer (2004) 40:667-72.
[205] Harmon AW, Patel YM. Naringenin inhibits glucose uptake in MCF-7 breast cancer
cells: a mechanism for impaired cellular proliferation. Breast Cancer Res Treat (2004) 85:103-10.
[206] Tannin-Spitz T, Bergman M, Grossman S. Cucurbitacin glucosides: antioxidant and
free-radical scavenging activities. Biochem Biophys Res Commun (2007) 364:181-6.
[207] Neeman M, Degani H. Metabolic studies of estrogen- and tamoxifen-treated human
breast cancer cells by nuclear magnetic resonance spectroscopy. Cancer Res (1989) 49:589-94.
[208] Forbes NS, Meadows AL, Clark DS, Blanch HW. Estradiol stimulates the biosynthetic
pathways of breast cancer cells: detection by metabolic flux analysis. Metab Eng (2006) 8:639-52.
[209] Singh A, Purohit A, Hejaz HA, Potter BV, Reed MJ. Inhibition of deoxyglucose uptake
in MCF-7 breast cancer cells by 2-methoxyestrone and 2-methoxyestrone-3-O-sulfamate. Mol Cell Endocrinol (2000) 160:61-6.
[210] Ben-Haim S, Ell P. 18F-FDG PET and PET/CT in the evaluation of cancer treatment
response. J Nucl Med (2009) 50:88-99.
[211] Goldin A. Ifosfamide in experimental tumor systems. Semin Oncol (1982) 9:14-23.
[212] Borner K, Kisro J, Bruggemann SK, Hagenah W, Peters SO, et al. Metabolism of
ifosfamide to chloroacetaldehyde contributes to antitumor activity in vivo. Drug Metab Dispos (2000) 28:573-6.
[213] Bruggemann SK, Radike K, Braasch K, Hinrichs J, Kisro J, et al. Chloroacetaldehyde:
mode of antitumor action of the ifosfamide metabolite. Cancer Chemother Pharmacol (2006) 57:349-56.
[214] Motrescu ER, Otto AM, Brischwein M, Zahler S, Wolf B. Dynamic analysis of
metabolic effects of chloroacetaldehyde and cytochalasin B on tumor cells using bioelectronic sensor chips. J Cancer Res Clin Oncol (2005) 131:683-91.
[215] Takahashi K, Sakurai K, Tanaka H, Fujimoto Y. Necrotic pathway in human
osteosarcoma Saos-2 cell death induced by chloroacetaldehyde. Anticancer Drugs (2007) 18:543-53.
[216] Trautschold I, Lamprecht W, Schweitzer G. ATP: UV-method with hexokinase and
glucose-6-P-dehydrogenase. In: Methods of enzymatic analysis, 3rd edn. Bergmeyer HU, editors. VCH Verlagsgesellschaft, Weinheim (1985). vol VII, pp 346-57.
[217] Kunst A, Draeger B, Ziegenhorn J. D-Glucose: UV-methods with hexokinase and
glucose-6-P-dehydrogenase. In: Methods of enzymatic analysis, 3rd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1984). vol VI, pp 163-72.
[218] Griffith OW. Glutathione and glutathione disulfide. In: Methods of enzymatic analysis,
3rd edn. Bergmeyer HU, editors. VCH Verlagsgesellschaft, Weinheim (1985). vol VIII, pp 521-9.
[219] Gutmann I, Wahlefeld AW. L-Lactate determination with lactate dehydrogenase and
NAD. In: Methods of enzymatic analysis, 2nd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1974). vol 3, pp 1464-8.
[220] Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin
phenol reagent. J Biol Chem (1951) 193:265-75.
[221] Lamprecht W, Heinz F. Pyruvate. In: Methods of enzymatic analysis, 3rd edn.
Bergmeyer HU, editors. Verlag Chimie, Weinheim (1984). vol VI, pp 570-7.
[222] Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys (1959) 82:70-7. [223] Sakai A, Shimizu H, Kono K, Furuya E. Monochloroacetic acid inhibits liver
gluconeogenesis by inactivating glyceraldehyde-3-phosphate dehydrogenase. Chem Res Toxicol (2005) 18:277-82.
[224] Passonneau JV, Lowry OH. Glucose-6-Phosphatase (EC 3139). In: Enzymatic
analysis: a practical guide. The Human Press, Totowa, New Jersey (1993). pp 252-4.
[225] Latzko E, Gibbs M. Alkaline C1-Fructose-1,6-disphosphatase. In: Methods of
enzymatic analysis, 2nd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1974). vol 2, pp 881-4.
[226] Passonneau JV, Lowry OH. Phosphoenolpyruvate Carboxykinase (EC 41132). In:
Enzymatic analysis: a practical guide. The Human Press, Totowa, New Jersey (1993). pp 291-2.
[227] Vassault A. Lactate dehydrogenase: UV-method with pyruvate and NADH. In:
Methods of enzymatic analysis, 3rd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1983). vol III, pp 118-26.
[228] Cain K, Skilleter DN. Chapter 9: Preparation and use of mitochondria in toxicological
research. In: Biochemical Toxicology: a practical approach, 1st edn. Snell K, editors. IRL Press, Oxford (1987). vol pp 217-54.
[229] Bergmeyer HU, Gawehn K, Grassl M. Enzymes as biochemical reagents: Fructose-6-
phosphate Kinase, Glucose-6-phosphate Dehydrogenase, Hexokinase. In: Methods of enzymatic analysis, 2nd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1974). vol 1, pp 451-74.
[230] Gutmann I, Bernt E. Pyruvate kinase. Assay in serum and erythrocytes. In: Methods
of enzymatic analysis, 2nd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1974). vol 2, pp 774-8.
[231] Michal G, Beutler HO. D-Fructose-1,6-diphosphate, Dihydroxyacetone phosphate and
D-Glyceraldehyde-3-phosphate. In: Methods of enzymatic analysis, 2nd edn. Bergmeyer HU, editors. Verlag Chemie, Weinheim (1974). vol 3, pp 1314-9.
[232] Parrish AR, Gandolfi AJ, Brendel K. Precision-cut tissue slices: applications in
pharmacology and toxicology. Life Sci (1995) 57:1887-901.
[233] Krumdieck CL, dos Santos JE, Ho KJ. A new instrument for the rapid preparation of
tissue slices. Anal Biochem (1980) 104:118-23.
[234] de Graaf IA, Draaisma AL, Schoeman O, Fahy GM, Groothuis GM, et al.
Cryopreservation of rat precision-cut liver and kidney slices by rapid freezing and vitrification. Cryobiology (2007) 54:1-12.
[235] Lerche-Langrand C, Toutain HJ. Precision-cut liver slices: characteristics and use for
in vitro pharmaco-toxicology. Toxicology (2000) 153:221-53.
[236] Vickers AE, Fisher RL. Organ slices for the evaluation of human drug toxicity. Chem
Biol Interact (2004) 150:87-96.
[237] Vittorelli A, Gauthier C, Michoudet C, Baverel G. Metabolic viability and pharmaco-
toxicological reactivity of cryopreserved human precision-cut renal cortical slices. Toxicol In Vitro (2004) 18:285-92.
[238] Richter E, Friesenegger S, Engl J, Tricker AR. Use of precision-cut tissue slices in
organ culture to study metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) by hamster lung, liver and kidney. Toxicology (2000) 144:83-91.
[239] Vittorelli A, Gauthier C, Michoudet C, Martin G, Baverel G. Characteristics of
glutamine metabolism in human precision-cut kidney slices: a 13C-NMR study. Biochem J (2005) 387:825-34.
[240] Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, et al. Enhancement of
chemotherapy by manipulation of tumour pH. Br J Cancer (1999) 80:1005-11.
[241] D'Souza RW, Francis WR, Andersen ME. Physiological model for tissue glutathione
depletion and increased resynthesis after ethylene dichloride exposure. J Pharmacol Exp Ther (1988) 245:563-8.
[242] Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol
Interact (1998) 111-112:1-14.
[243] Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am J Physiol (1989)
[244] Rick O, Beyer J, Schwella N, Schubart H, Schleicher J, et al. Assessment of
amifostine as protection from chemotherapy-induced toxicities after conventional-dose and high-dose chemotherapy in patients with germ cell tumor. Ann Oncol (2001) 12:1151-5.
[245] Utter MF, Keech DB. Formation of oxaloacetate from pyruvate and carbon dioxide. J
Biol Chem (1960) 235:PC17-8.
[246] Madsen KM, Tisher CC. Structural-functional relationship along the distal nephron.
Am J Physiol (1986) 250:F1-15.
[247] Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int (1984)
[248] Trentham DR. Aspects of the chemistry of D-glyceraldehyde 3-phosphate
dehydrogenase. Biochem J (1968) 109:603-12.
[249] Wolny M, Banas B, Banas T. Human muscle glyceraldehyde-3-phosphate
dehydrogenase: reactivity of sulphydryl groups. Acta Biochim Pol (1982) 29:189-96.
[250] Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-
induced endothelial dysfunction. Biochim Biophys Acta (2006) 1760:267-71.
[251] Pullar JM, Winterbourn CC, Vissers MC. Loss of GSH and thiol enzymes in
endothelial cells exposed to sublethal concentrations of hypochlorous acid. Am J Physiol (1999) 277:H1505-12.
[252] Dhanotiya RS, Bhardwaj R. Properties of renal fructose 1,6 bis-phosphatase of goat.
Arch Exp Veterinarmed (1990) 44:395-9.
[253] Reyes A, Burgos ME, Hubert E, Slebe JC. Selective thiol group modification renders
fructose-1,6-bisphosphatase insensitive to fructose 2,6-bisphosphate inhibition. J Biol Chem (1987) 262:8451-4.
[254] Reyes A, Hubert E, Slebe JC. The reactive cysteine residue of pig kidney fructose
1,6-bisphosphatase is related to a fructose 2,6-bisphosphate allosteric site. Biochem Biophys Res Commun (1985) 127:373-9.
[255] Liu F, Fromm HJ. Relationship between thiol group modification and the binding site
for fructose 2,6-bisphosphate on rabbit liver fructose-1,6-bisphosphatase. J Biol Chem (1988) 263:10035-9.
[256] Clottes E, Burchell A. Three thiol groups are important for the activity of the liver
microsomal glucose-6-phosphatase system. Unusual behavior of one thiol located in the glucose-6-phosphate translocase. J Biol Chem (1998) 273:19391-7.
[257] Clottes E, Middleditch C, Burchell A. Rat liver glucose-6-phosphatase system: light
scattering and chemical characterization. Arch Biochem Biophys (2002) 408:33-41.
[258] Soares JC, Folmer V, Rocha JB. Influence of dietary selenium supplementation and
exercise on thiol-containing enzymes in mice. Nutrition (2003) 19:627-32.
[259] Lewis CT, Seyer JM, Carlson GM. Cysteine 288: an essential hyperreactive thiol of
cytosolic phosphoenolpyruvate carboxykinase (GTP). J Biol Chem (1989) 264:27-33.
[260] Dupuis A, Skehel JM, Walker JE. NADH:ubiquinone oxidoreductase from bovine
mitochondria. cDNA sequence of a 19 kDa cysteine-rich subunit. Biochem J (1991) 277 ( Pt 1):11-5.
[261] Balijepalli S, Annepu J, Boyd MR, Ravindranath V. Effect of thiol modification on brain
mitochondrial complex I activity. Neurosci Lett (1999) 272:203-6.
[262] Sriram K, Shankar SK, Boyd MR, Ravindranath V. Thiol oxidation and loss of
mitochondrial complex I precede excitatory amino acid-mediated neurodegeneration. J Neurosci (1998) 18:10287-96.
[263] Sehirli O, Sakarcan A, Velioglu-Ogunc A, Cetinel S, Gedik N, et al. Resveratrol
improves ifosfamide-induced Fanconi syndrome in rats. Toxicol Appl Pharmacol (2007) 222:33-41.
[264] Salahudeen AK. Role of lipid peroxidation in H2O2-induced renal epithelial (LLC-
PK1) cell injury. Am J Physiol (1995) 268:F30-8.
[265] Sheridan AM, Fitzpatrick S, Wang C, Wheeler DC, Lieberthal W. Lipid peroxidation
contributes to hydrogen peroxide induced cytotoxicity in renal epithelial cells. Kidney Int (1996) 49:88-93.
[266] Kumagai S, Narasaki R, Hasumi K. Glucose-dependent active ATP depletion by
koningic acid kills high-glycolytic cells. Biochem Biophys Res Commun (2008) 365:362-8.
[267] Mazurek S, Grimm H, Wilker S, Leib S, Eigenbrodt E. Metabolic characteristics of
different malignant cancer cell lines. Anticancer Res (1998) 18:3275-82.
[268] Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor
model for liver cancer: characterization and targeting hexokinase. Cancer Lett (2001) 173:83-91.
[269] Lee YJ, Galoforo SS, Berns CM, Tong WP, Kim HR, et al. Glucose deprivation-
induced cytotoxicity in drug resistant human breast carcinoma MCF-7/ADR cells: role of c-myc and bcl-2 in apoptotic cell death. J Cell Sci (1997) 110 ( Pt 5):681-6.
[270] Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic
pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J (2002) 364:309-15.
[271] Ramos-Montoya A, Lee WN, Bassilian S, Lim S, Trebukhina RV, et al. Pentose
phosphate cycle oxidative and nonoxidative balance: A new vulnerable target for overcoming drug resistance in cancer. Int J Cancer (2006) 119:2733-41.
[272] Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy
metabolism in tumor cells. FEBS J (2007) 274:1393-418.
[273] Ursini MV, Parrella A, Rosa G, Salzano S, Martini G. Enhanced expression of
glucose-6-phosphate dehydrogenase in human cells sustaining oxidative stress. Biochem J (1997) 323 ( Pt 3):801-6.
[274] Pereira da Silva AP, El-Bacha T, Kyaw N, dos Santos RS, da-Silva WS, et al.
Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem J (2009) 417:717-26.
[275] Brown JM. The hypoxic cell: a target for selective cancer therapy-eighteenth Bruce F.
Cain Memorial Award lecture. Cancer Res (1999) 59:5863-70.
[276] Guppy M. The hypoxic core: a possible answer to the cancer paradox. Biochem
Biophys Res Commun (2002) 299:676-80.
[277] Li J, Shi M, Cao Y, Yuan W, Pang T, et al. Knockdown of hypoxia-inducible factor-
1alpha in breast carcinoma MCF-7 cells results in reduced tumor growth and increased sensitivity to methotrexate. Biochem Biophys Res Commun (2006) 342:1341-51.
Source: http://researches.albaath-univ.edu.sy/archive/books/%D9%84%D9%85%20%D8%A3%D8%AC%D8%AF%20%D9%84%D9%87%D9%85%20%D8%B1%D9%82%D9%85%20%D8%B9%D9%84%D9%89%20%D8%A7%D9%84%D9%85%D9%88%D9%82%D8%B9/%D8%B7%D8%A8%20%D8%A8%D8%B4%D8%B1%D9%8A/%D8%A3%D8%B7%D8%B1%D9%88%D8%AD%D8%A9%20%D8%A8%D8%B1%D9%87%D8%A7%D9%86%20%D9%83%D9%86%D9%88%D8%B2%D9%8A.pdf
Free trade spanish jh
Libre comercio para mejor saludPhilip StevensPrefacio por Dr Rigoberto Stewart, Director del Instituto para la Libertad y el Análisis de Políticas, Costa Rica Libre comercio paramejor salud Prefacio por Dr Rigoberto Stewart,Director del Instituto para la Libertad y el Análisis de Políticas, Costa Rica Libre comercio para mejor salud
Edc7b.qxd
Traitements anti-parasitaires du bétail, insectes coprophages & chauves-souris Avertissement :ce dossier complexe,sujet très controversé au sein de la communauté des scientifiques,spécialistes du sujet,n'a qu'un seul objectif,celui de vous apporter un maximum d'informations sur la problématique «traitements anti-parasitaires».Les solutions proposées sont