Superresolution imaging of human cytomegalovirus vmia localization in sub-mitochondrial compartments
Viruses 2014, 6, 1612-1636; doi:10.3390/v6041612OPEN ACCESS
ISSN 1999-4915
Superresolution Imaging of Human Cytomegalovirus vMIA
Localization in Sub-Mitochondrial Compartments
Shivaprasad Bhuvanendran 1, Kyle Salka 1, Kristin Rainey 2, Sen Chandra Sreetama 1,
Elizabeth Williams 1, Margretha Leeker 1, Vidhya Prasad 1, Jonathan Boyd 3,
George H. Patterson 2,*, Jyoti K. Jaiswal 1,4,* and Anamaris M. Colberg-Poley 1,4,5,*
1 Research Center for Genetic Medicine, Children's Research Institute, Children's National Health System, 111 Michigan Avenue, NW, Washington, DC 20010, USA; E-Mails: [email protected] (S.B.); [email protected] (K.S.); [email protected] (S.C.S.); [email protected] (E.W.); [email protected] (M.L.); [email protected] (V.P.) 2 Section on Biophotonics, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, MD 20892, USA; E-Mail: [email protected] 3 Life Science Division, Leica Microsystems, Inc., 1700 Leider Lane, Buffalo Grove, IL 60089, USA; E-Mail: [email protected] 4 Department of Integrative Systems Biology, George Washington University School of Medicine and Health Sciences, Washington, DC 20037, USA 5 Department of Biochemistry and Molecular Medicine, George Washington University School of Medicine and Health Sciences, Washington, DC 20037, USA * Authors to whom correspondence should be addressed; E-Mails: [email protected] (G.H.P.);
[email protected] (J.K.J.); [email protected] (A.M.C.-P.); Tel.: +1-202-476-3984 (A.M.C.-P.); Fax: +1-202-476-6014 (A.M.C.-P.). Received: 17 January 2014; in revised form: 16 March 2014 / Accepted: 27 March 2014 / Published: 9 April 2014 Abstract: The human cytomegalovirus (HCMV) viral mitochondria-localized inhibitor of
apoptosis (vMIA) protein, traffics to mitochondria-associated membranes (MAM), where
the endoplasmic reticulum (ER) contacts the outer mitochondrial membrane (OMM).
vMIA association with the MAM has not been visualized by imaging. Here, we have
visualized this by using a combination of confocal and superresolution imaging.
Deconvolution of confocal microscopy images shows vMIA localizes away from
mitochondrial matrix at the Mitochondria-ER interface. By gated stimulated emission
Viruses 2014, 6
depletion (GSTED) imaging, we show that along this interface vMIA is distributed in clusters. Through multicolor, multifocal structured illumination microscopy (MSIM), we find vMIA clusters localize away from MitoTracker Red, indicating its OMM localization. GSTED and MSIM imaging show vMIA exists in clusters of 100–150 nm, which is consistent with the cluster size determined by Photoactivated Localization Microscopy (PALM). With these diverse superresolution approaches, we have imaged the clustered distribution of vMIA at the OMM adjacent to the ER. Our findings directly compare the relative advantages of each of these superresolution imaging modalities for imaging components of the MAM and sub-mitochondrial compartments. These studies establish the ability of superresolution imaging to provide valuable insight into viral protein location, particularly in the sub-mitochondrial compartments, and into their clustered organization. Keywords: HCMV vMIA; MAM; mitochondria; OMM; matrix; confocal microscopy;
superresolution microscopy; GSTED; MSIM; PALM
1. Introduction
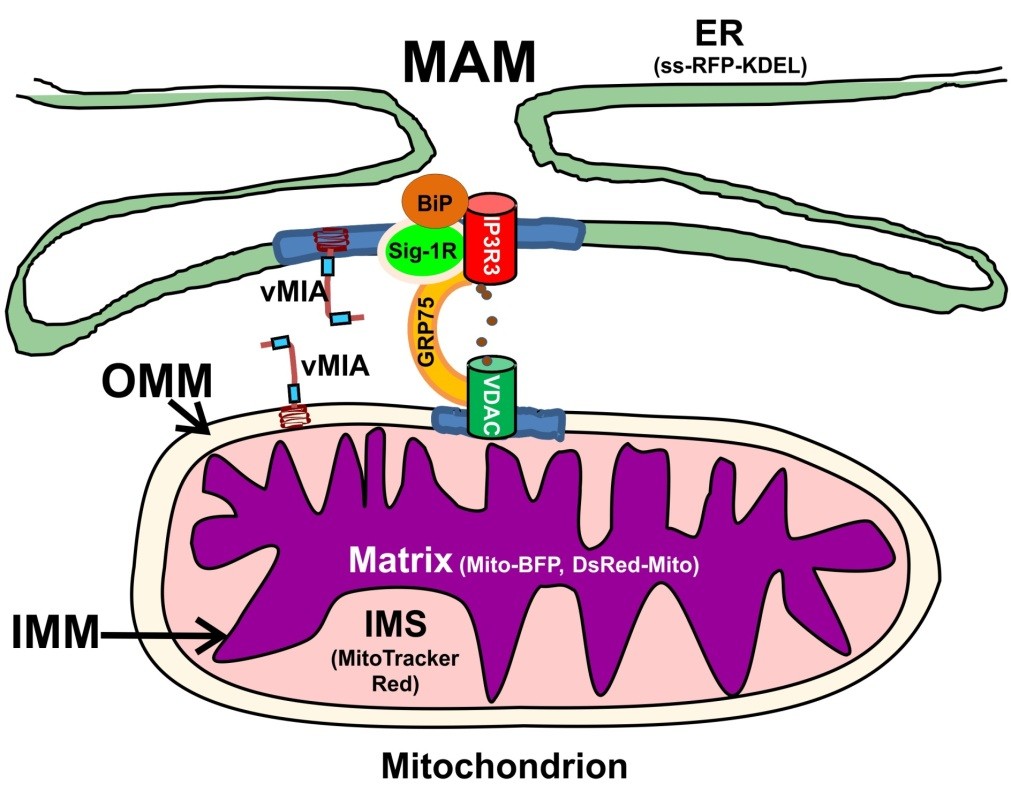
The mitochondria-associated membrane (MAM) sub-compartment of the endoplasmic reticulum (ER) plays critical roles in ER-mitochondrial cross-talk by allowing efficient transfer of calcium (Ca2+) from the ER to mitochondria without elevating cytosolic Ca2+ levels [1–3]. A macromolecular complex, composed of MAM inositol 1,4,5 trisphosphate receptors (IP3R), cytosolic glucose response protein 75 (GRP75) and outer mitochondrial membrane (OMM)-localized voltage dependent anion channel (VDAC) generates the high Ca2+ microdomains needed for Ca2+ transfer from ER to mitochondria (Figure 1) [4]. Constitutive low level IP3R-mediated Ca2+ transfer, needed for Ca2+ dependent mitochondrial enzymes, maintains normal cellular metabolism [5]. Nonetheless, continued mitochondrial Ca2+ influx drives the adaptive metabolic phase of early ER stress and can result in mitochondrial-mediated apoptosis [6,7]. Lipids, including phospholipids, cholesterol, and ceramide, are synthesized in the MAM and transferred to the OMM [1,8,9]. In addition, the MAM is enriched in internal lipid rafts [10], which can serve to connect extrinsic and intrinsic apoptotic pathways [11]. Finally, mitochondrial antiviral responses have also been recently linked to the MAM [7,12,13]. The viral mitochondria-localized inhibitor of apoptosis (vMIA), encoded by the human cytomegalovirus (HCMV) UL37 exon 1 (UL37x1) immediate early gene, inhibits mitochondrial- mediated programmed cell death and increases viral progeny production during permissive infection [14–19]. vMIA traffics sequentially from the ER to mitochondria and is present at ER-mitochondria contact sites known as MAM [20–24]. At the ER, vMIA causes ER Ca2+ efflux [15]. It associates with MAM lipid rafts in close proximity to sigma 1 receptor (Sig-1R) [25], which is a chaperone affecting Ca2+ efflux from the ER. Additionally, vMIA recruits Bax to MAM lipid rafts and induces Bax proteasome-mediated degradation, thereby augmenting vMIA's antiapoptotic activity [26,27]. Because of its sequential trafficking [20,23], vMIA can relocalize a cellular defense protein, viperin, from the ER to mitochondria where viperin assumes a new role of a major effector to induce lipogenesis metabolism during HCMV infection [28,29]. At mitochondria, vMIA blocks Bax-mediated
Viruses 2014, 6
permeabilization of the OMM [17,18,30,31], reduces ATP synthesis [28,29,32], causes mitochondrial
fragmentation [12,30,33,34], and controls HtrA2/Omi-induced cell death through very late times of
HCMV infection [35].
Figure 1. Endoplasmic reticulum (ER), mitochondria-associated membranes (MAM), and
mitochondrial sub-compartments visualized. Viral mitochondria-localized inhibitor of
apoptosis (vMIA) localization in the MAM and mitochondrion sub-compartments was
imaged using the following markers: preprolactin signal sequence (ss) fused to the
N-terminus of the red fluorescent protein (RFP) with a KDEL ER retention signal at its
C-terminus (ss-RFP-KDEL, for ER) [36] and mitochondrial Cox 4 leader fused to Tag blue
fluorescent protein (Mito-BFP)/mitochondrial targeting sequence from human cytochrome
c oxidase subunit VIII to the N-terminus of Discosoma RFP (DsRed-Mito, for mitochondrial
matrix) [37]. We use MitoTracker Red as an intermembrane space (IMS) marker based
upon the superresolution imaging of MitoTracker by others [38,39] and our own results
herein. Contacts between the ER and mitochondria are shown, with the MAM Ca2+
signaling complex components on the ER (IP3R3), cytosol (GRP75) and outer
mitochondrial membrane (OMM) (voltage dependent anion channel (VDAC)). MAM Ca2+
efflux from the ER is regulated by chaperones (BiP, Sig-1R) as well as vMIA. Lipid rafts
(blue) containing the Ca2+ signaling complex and vMIA are shown. These components are
shown in the figure.
vMIA is N-terminally anchored to ER and mitochondrial membranes by an uncleaved hydrophobic
leader and its downstream C-terminal sequences are cytosolic [20]. This topology was confirmed by
Viruses 2014, 6
vMIA's sensitivity to protease digestion in ER and mitochondrial fractions [22]. Immune electron
microscopy (EM) of stably transfected HeLa cells has localized vMIA-myc using anti-myc antibody
primarily at the OMM [16]. However, fixation, embedding and staining of specimens for EM severely
compromise membrane morphology. Moreover, immune EM limits identifying the distribution of the
protein populations such that only a fraction of the molecules in a given cellular organelle can be
detected, thereby offering limited information about spatial distribution of the targeted molecule. Thus,
the exact pattern of vMIA distribution and functional organization along the mitochondria was not
detected by immune EM analysis. This EM analysis aside, vMIA imaging has primarily utilized
conventional confocal microscopy [15–18,20,23,24,30–32,35,40]. Using multicolor confocal microscopy,
we have previously found that enhanced green fluorescent protein (EGFP) tagged vMIA partially
co-localizes with MAM, lipid raft, and mitochondrial markers [20,21,23]. Further, vMIA has been
co-localized with mitochondrial markers from the OMM and matrix [17,18,20,21,23–25,32].
A major challenge in precisely defining vMIA's localization in the ER, MAM and sub-mitochondrial
compartments by confocal microscopy results from the close proximities of the ER and OMM membranes (10–25 nm) and of the OMM and inner mitochondrial membrane (IMM) at the MAM, which are below its diffraction limit. While confocal microscopy can theoretically produce a resolution down to 200 nm, this resolution of visible light is seldom achieved in practice due to numerous optical aberrations associated with biological specimens as well as noise associated with the detected fluorescence. Some of this can be corrected by deconvolution of confocal images [41]. Mitochondria typically have a diameter of 200–500 nm [38]. Thus, it is not possible to determine vMIA distribution within sub-mitochondrial compartments using conventional confocal microscopy. For this, we turned to superresolution microscopy, which allows imaging beyond the limitations imposed by diffraction, to improve insight into vMIA's distribution in sub-mitochondrial compartments.
Superresolution microscopy overcomes the physical limit imposed by diffraction. Multiple
approaches have been developed to resolve fluorescent signals below diffraction limit and these include structured illumination (e.g., structured illumination microscopy, SIM; multifocal SIM, MSIM), reduction of point spread function by grounding emissions outside of the excitation center (e.g., stimulated emission depletion, STED; gated STED, GSTED) or activation of single fluorophores (e.g., photoactivated localization microscopy, PALM; stochastic optical reconstruction microscopy, STORM). Each of these approaches has its own strengths and weaknesses. In this study, we examined the localization of vMIA by deconvolved confocal microscopy and three superresolution microscopy techniques namely MSIM, single-color GSTED, and PALM.
MSIM uses sparse 2D excitation patterns [42] moved in sequential steps to fully illuminate the
specimen. Images are collected at each step and used in post processing to derive a superresolution image. Superresolution is achieved by first defining the precise location of the illumination spots. Once the focal spots in each of the images are defined, these are digitally pinholed followed by scaling the spots by a factor of 0.5 and then summing these images over all positions. The pinholed, scaled, and summed images are then subjected to Richardson-Lucy deconvolution to gain 2-fold improvement in resolution. Although MSIM sacrifices speed compared to confocal microscopy, it maintains the optical sectioning and was shown to provide resolution-doubling characteristics of SIM to 140 nm [42].
Further improvement in resolution over MSIM is obtained using STED microscopy, where a
592 nm wavelength doughnut shaped beam is used to drive the fluorochromes in the doughnut to
Viruses 2014, 6
ground state by stimulated emission resulting in <50 nm resolution [43,44]. Time gating of the short
fluorescent lifetimes caused by stimulated emission results in the GSTED approach further improves
the spatial resolution. STED microscopy has previously shown that the mitochondrial inner membrane
organizing system (MINOS) forms clusters within mitochondria of primary human fibroblasts [45]
while two color STED found VDAC type 3 and hexokinase I clusters on the OMM of human
osteosarcoma cells [46]. Similarly, cytochrome c oxidase subunit 2 and VDAC1 were found in clusters
in purified mitochondria from murine heart [47].
Pointillistic imaging based superresolution microscopy approach, PALM offers the highest
resolution microscopy ( 25 nm) used in these studies. PALM uses photoactivatable fluorescent proteins and precise localization of single molecules to overcome diffraction limitations. PALM is based on high density, single molecule localization in which single molecule signals are fitted with 2D Gaussian functions to provide a more precise estimate of the molecule's location. PALM and several related techniques use photoactivatable, photoswitchable, or photoconvertible fluorescent proteins [48], which initially have little fluorescence or their fluorescence can be turned ―off‖ in the spectral region under detection before they are actively turned ―on‖ during imaging. Conservatively setting the precision cutoff at 50 nm can often produce images resolved at that value which is 3-fold improvement over the MSIM images and comparable to the GSTED images. Similar to MSIM, PALM provides the ability to image multiple colors and better resolution than confocal, but slower imaging speed.
2. Results and Discussion
2.1. Conventional Confocal Imaging of vMIA Localization with Mitochondrial Markers
We first used confocal microscopy to monitor mitochondrial distribution of vMIA in HCMV
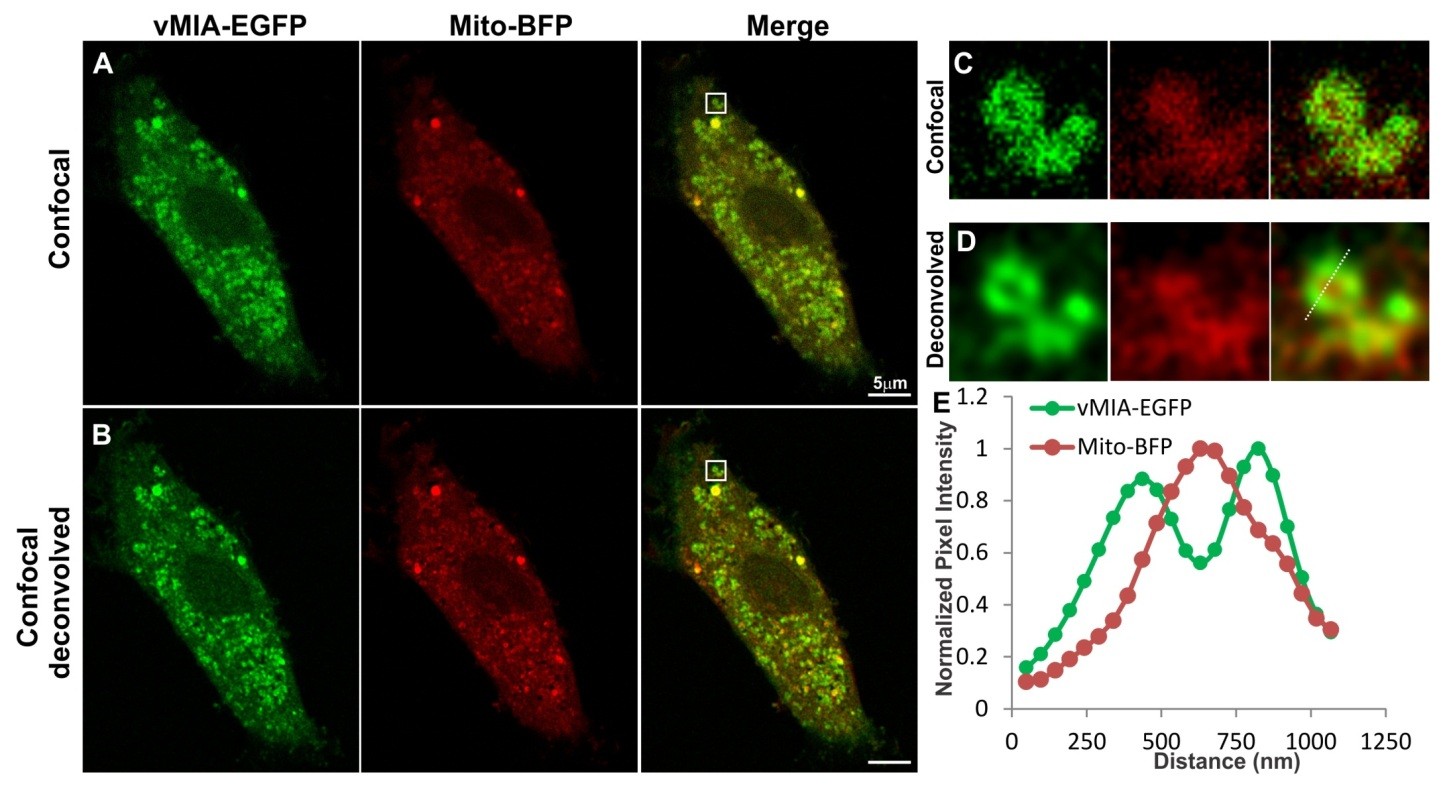
permissive cells, human foreskin fibroblasts (HFFs) expressing vMIA-EGFP, which traffics indistinguishably from untagged vMIA [15,21,23,26]. We labeled the mitochondrial matrix by using S. cerevisiae mitochondrial Cox 4 leader fused to Tag blue fluorescent protein (Mito-BFP) [37] (Figure 2). As we previously found [20,21,23,24], vMIA-EGFP fluorescence substantially co-localized with a matrix marker, Mito-BFP [37] (Figure 2A). The zoom of one of the larger mitochondrion (Figure 2C) shows an example of the level of detail available when imaging these markers with confocal microscopy. With the spatial resolution we obtained for this image (FWHM for vMIA-EGFP = 298 nm), vMIA-EGFP distribution was only marginally distinguishable from Mito-BFP. With the image acquired 3-times below the Nyquist limit, deconvolution of the confocal Z-stack images improved the spatial resolution (FWHM for vMIA-EGFP = 182 nm) (Figure 2B). This improved resolution allowed us to distinguish the presence of vMIA at the rim of the mitochondria, away from the matrix marker Mito-BFP (Figure 2D). This is quantified by the intensity profile for the dotted line marked along the deconvolved image of the mitochondrion (Figure 2E). This imaging also demonstrated that vMIA is not uniformly distributed along the mitochondrial periphery, hinting at the possibility of clustering of vMIA on the mitochondrial surface (Figure 2D); however, the clusters of molecules were not clearly resolvable even after deconvolution of the confocal microscopy images. In
Viruses 2014, 6
summary, confocal microscopy showed the presence of vMIA at the periphery of mitochondria and
partially resolved it from the matrix marker.
Figure 2. Monitoring mitochondrial localization of vMIA by confocal microscopy.
Primary human foreskin fibroblasts (HFFs) lipofected with vectors expressing vMIA-
EGFP and Mito-BFP were fixed with 4% paraformaldehyde (PFA) at 22 hours after
transfection as described in the methods. (A) Images show a single optical plane for a cell
expressing vMIA-EGFP (green) and Mito-BFP (pseudocolored red) imaged using confocal
microscopy and (B) the same image plane following deconvolution of the entire Z-stack.
(C,D) The boxed region of interest is enlarged on the right. (E) Intensity profile of
vMIA-EGFP (green) and Mito-BFP (red) emissions along the pixels marked by the dotted
line on the deconvolved image are shown by the plot. For higher resolution images, see
Supplemental Figure S1.
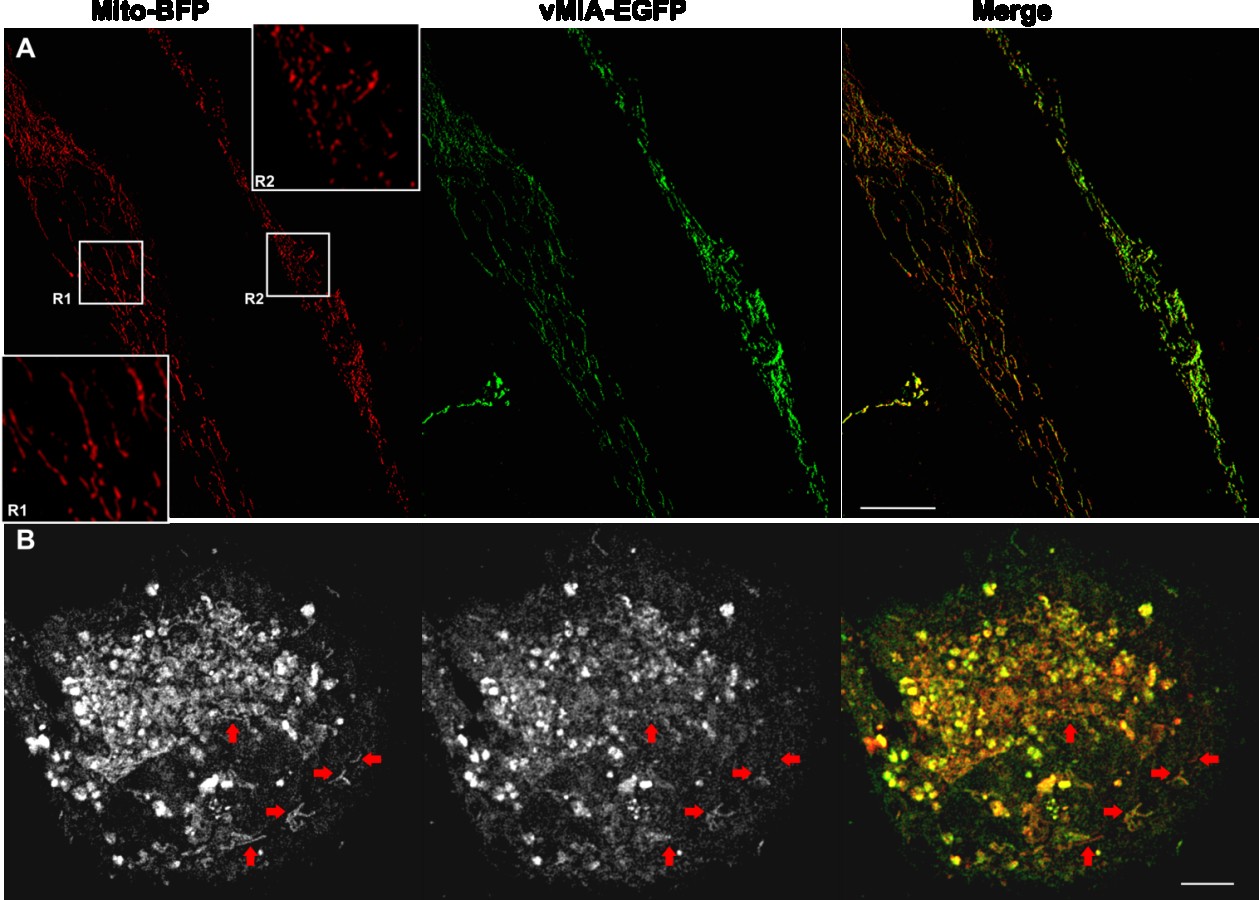
In these studies, we detected altered mitochondrial morphology in HFFs expressing vMIA
consistent with previous literature [30,33,34,49]. To determine if the altered mitochondrial morphology correlated with vMIA levels, we examined mitochondria morphology in cells expressing vMIA (Figure 3). We found that mitochondria (red) not expressing vMIA or expressing low levels of vMIA (R1) maintained tubular morphology (Figure 3A). Conversely, mitochondria expressing higher levels of vMIA (R2) showed fragmented, vesicular morphology. Multiple tubular mitochondria (blue arrows) not expressing or expressing low levels of vMIA were also observed in another cell (Figure 3B). These results suggest that threshold levels of vMIA are required for mitochondrial fragmentation and vesiculation.
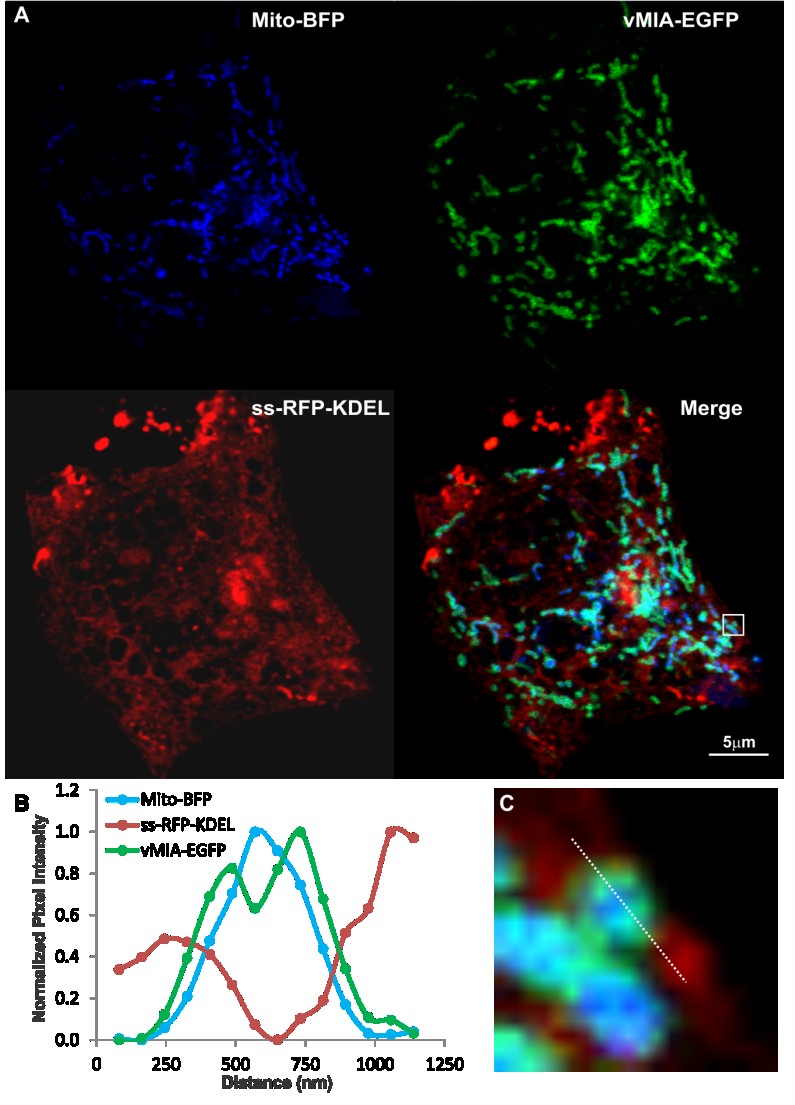
With the ability of the deconvolved confocal image to resolve vMIA distribution from the
mitochondrial matrix marker (Mito-BFP), we next examined the distribution of these markers with respect to that of the ER (ss-RFP-KDEL), which has the bovine preprolactin signal sequence (ss) fused
Viruses 2014, 6
to monomeric red fluorescent protein (RFP) with the KDEL ER retention [36] sequence (graciously
provided by Dr. J. Lippincott-Schwartz) (Figure 4A). HFFs expressing the three fluorophore-tagged
proteins showed that vMIA partially colocalized with ER and mitochondrial markers as we previously
found [20,21,23,24,26,50]. Moreover, the intensity profile (Figure 4B) of the pixels along the line
shown in the zoomed boxed region of interest (Figure 4C) showed that vMIA-EGFP is located at the
interface of Mito-BFP and ss-RFP-KDEL. Although there is detection of the interfaces between the ER
and mitochondria, there is substantial overlap of the mitochondrial OMM (green) and matrix (blue)
sub-compartments, which limits the ability of confocal microscopy to compellingly resolve the
sub-mitochondrial distribution of vMIA.
Figure 3. Mitochondrial fragmentation by vMIA exhibits a threshold effect. Primary
HFFs transfected with vectors to express vMIA-EGFP and Mito-BFP were fixed with 4%
PFA at 22 hours after transfection as described in the methods and visualized by confocal
deconvolution microscopy. (A) Maximal intensity projection of a 3-D image showing a
pair of cells expressing differing levels of vMIA-EGFP (green), but comparable level of
Mito-BFP (pseudocolored red). Insets showing the zoom of regions R1 and R2 highlights
the change in mitochondrial morphology (as shown by Mito-BFP) in cells expressing low
level (R1) or high level (R2) of vMIA-GFP (B) Maximal intensity 3-D projection of
another cell expressing Mito-BFP (pseudocolored red in the merge) and showing varying
levels of vMIA-EGFP (green in the merge) on the individual mitochondria. While most
of the mitochondria in this cell have lost their tubular appearance, red arrows point to the
individual mitochondrion that show low or no vMIA-GFP expression have remained
tubular. Scale bars represent 5 µm. For higher resolution images, see Supplemental
Figure S2.
Viruses 2014, 6
Figure 4. Confocal microscopy imaging of ER-mitochondria interface. HFFs were
lipofected with vectors expressing vMIA-EGFP (OMM), ss-RFP-KDEL (ER) and
Mito-BFP (matrix) and fixed with 4% PFA at 25 hours after transfection as described
below and previously published [23]. (A) Cells expressing vMIA-EGFP (green),
ss-RFP-KDEL (red) and Mito-BFP (blue) were imaged using confocal microscopy.
(C) The boxed region of interest from the merged image is enlarged. (B) Line scans of
vMIA-EGFP (green), ss-RFP-KDEL (red) and Mito-BFP (blue) emissions are shown. For
higher resolution images, see Supplemental Figure S3.
Viruses 2014, 6
2.2. Single Color GSTED Imaging of vMIA Localization
To image vMIA beyond the diffraction limit imposed by the visible light, we used GSTED, an
earlier variant, STED, has been used to study clustered distribution of several mitochondrial proteins at the OMM and IMM by immune localization of intact or isolated mitochondria [45–47]. As use of antibody affects precision of localization due to increased distance added on by the presence of primary and fluorescently tagged secondary antibodies and imaging isolated mitochondria provides localization outside the biologically relevant subcellular context, we undertook in situ imaging of vMIA-EGFP in permissive HFFs (Figure 5A). Following deconvolution of the GSTED image, we detected vMIA-EGFP (green) in the periphery of a tubular mitochondrion (Figure 5B), which is consistent with the above suggestion of OMM localization by the confocal microscopy and of biochemical literature [16,20,22]. Intensity profile of pixels marked by the line shown in the zoomed GSTED image of the region R2 confirmed that vMIA is localized at the periphery of mitochondria, distinguishable from DsRed-Mito, used as a matrix marker and imaged by confocal microscopy (Figure 5D,E). GSTED showed improved resolution of vMIA (FWHM = 75 nm) OMM location compared to confocal imaging of vMIA (Figure 5B). This increase in spatial resolution also offered conclusive evidence to support clustered distribution of vMIA along the OMM (Figure 5C). Thus, superresolution GSTED resolved the presence of vMIA at the OMM and its clustered distribution in mitochondria in permissive HFFs. It also showed that vMIA exists as <100 nm clusters at the OMM irrespective of if the mitochondrion is tubular (Figure 5B) or fragmented (Figure 5D).
As clustering of vMIA along the OMM is an unprecedented phenotype for a viral protein, we
performed GSTED imaging of vMIA-EGFP in another permissive (human astrocytoma) cell line, U373-Tet-ON cells [51] (Figure 6A). GSTED imaging detected vMIA-EGFP (green) in U373-Tet-ON cells also showed that similar to HFFs, vMIA is present at the rim of the mitochondria (Figure 6B). This rim-like distribution of vMIA-EGFP was confirmed by the intensity profile of pixels marked by the line shown in the zoomed region (R2, Figure 6B). GSTED showed improved resolution of vMIA location (<50 nm) compared to confocal imaging (200 nm) (Figure 6C). Similar to our findings in HFFs (Figure 5), vMIA was present in clusters on the mitochondria in transfected U373-Tet-ON cells (Figure 6D,E). However, mitochondria of the U373-Tet-ON cells expressing vMIA are considerably less tubular (Figure 6) than mitochondria from transfected HFFs expressing vMIA (Figure 5). These differences in mitochondrial morphology likely represent cell-type specific differences in the effects of vMIA, which is known to alter mitochondrial morphology [30,32,33,52].
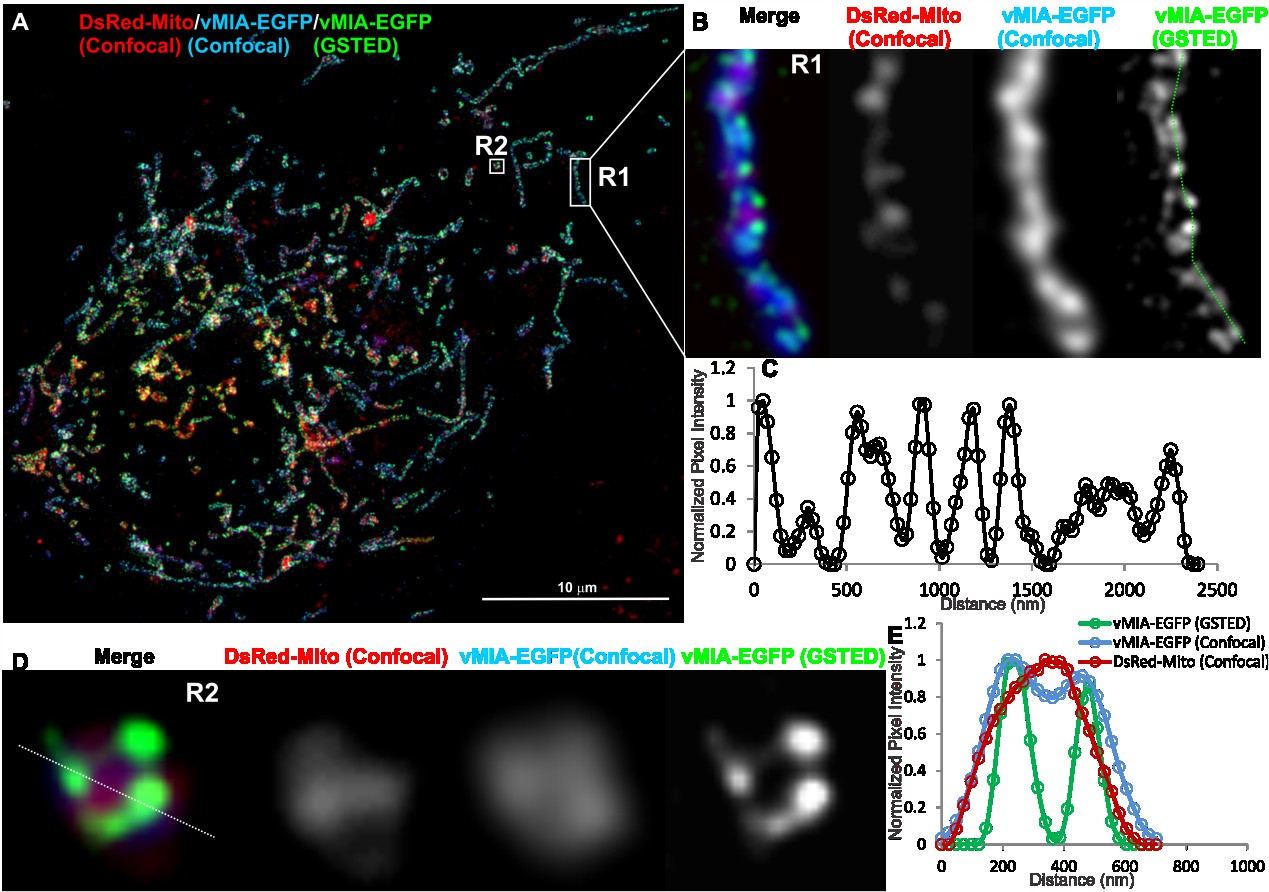
Viruses 2014, 6
Figure 5. Gated stimulated emission depletion (GSTED) microscopy of vMIA-EGFP in
human fibroblasts. (A) HFFs were lipofected with vectors expressing vMIA-EGFP and
DsRed-Mito. At 24 hours post transfection, cells were methanol fixed as described as
below and elsewhere [23] and imaged using GSTED (vMIA-EGFP) and confocal
microscopy (DsRed-Mito, vMIA-EGFP) followed by deconvolution of both the images.
(B) Zoomed, merged image of a tubular mitochondrion in the boxed region (R1) is shown.
This includes DsRed-Mito confocal (red), vMIA-EGFP confocal (blue) and vMIA-EGFP
GSTED (green). Each channel is also presented individually. (C) Intensity profile of the
pixels marked by the dotted line on the GSTED panel demonstrates the clustered
distribution of vMIA along the entire length of the OMM of this mitochondrion. (D) The
zoomed, merged image of a mitochondrion in the boxed region (R2) is shown. (E) The
normalized intensity profile along the line shown on the R2 image, which demonstrates the
significant improvement in visualizing the vMIA distribution along OMM by GSTED as
compared to confocal imaging and its improved resolution of its localization away from the
matrix and at the OMM. For higher resolution images, see Supplemental Figure S4.
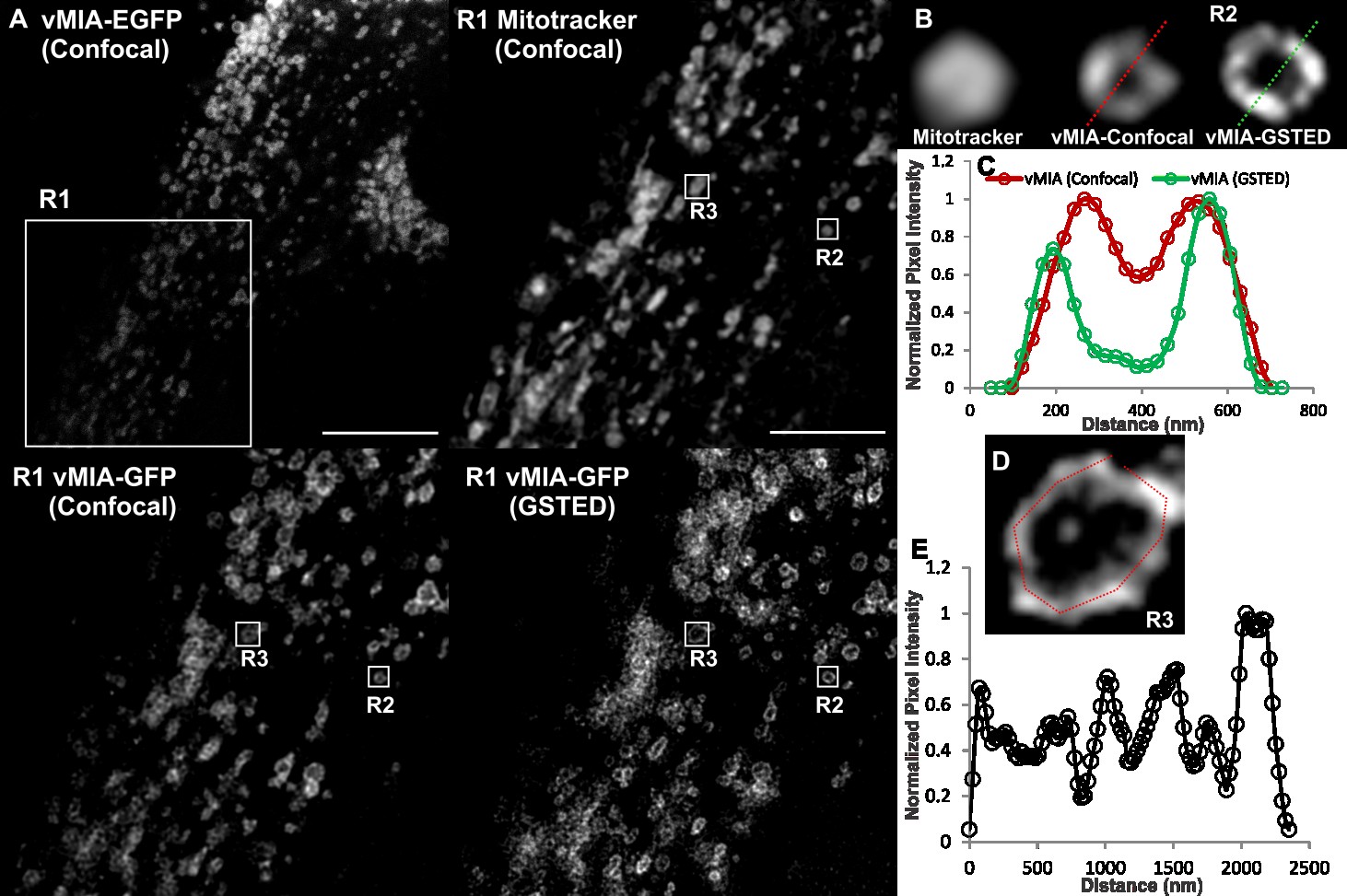
Viruses 2014, 6
Figure 6. GSTED microscopy of vMIA-EGFP in human astrocytoma cells. U373-Tet-ON
cells expressing the tetracycline controlled transactivator (tTA) [51] were lipofected
with the tetracycline responsive element (TRE-Tight) promoter driving expression of
vMIA-EGFP. At 24 hours post transfection, cells were treated with doxycycline (Dox) for
60 minutes, labeled with MitoTracker Red (0.5 µM) and imaged live using Confocal
(vMIA-GFP and MitoTracker Red) and GSTED (vMIA-GFP). (A) An optical slice
showing deconvolved confocal image of a cell expressing vMIA-EGFP and a zoom of the
region corresponding to the boxed region (R1) are shown in the left panel. Right panel
presents the zoom of the R1 region showing the deconvolved GSTED (bottom) and
deconvolved confocal MitoTracker (top) channels. (B) Zoomed confocal images of the
mitochondrion in the region R2 showing the various channels acquired—Confocal images
of MitoTracker Red and vMIA-EGFP as well as GSTED image of vMIA-EGFP.
(C) Normalized intensity profile along the dotted line shown on the confocal and GSTED
images of R2 demonstrate the improved resolution of vMIA localization by GSTED
imaging as compared to confocal imaging. (D) Zoom of region R3 showing GSTED image
of vMIA (E) Intensity profile of the pixels marked by the red line in panel (D). For higher
resolution images, see Supplemental Figure S5.
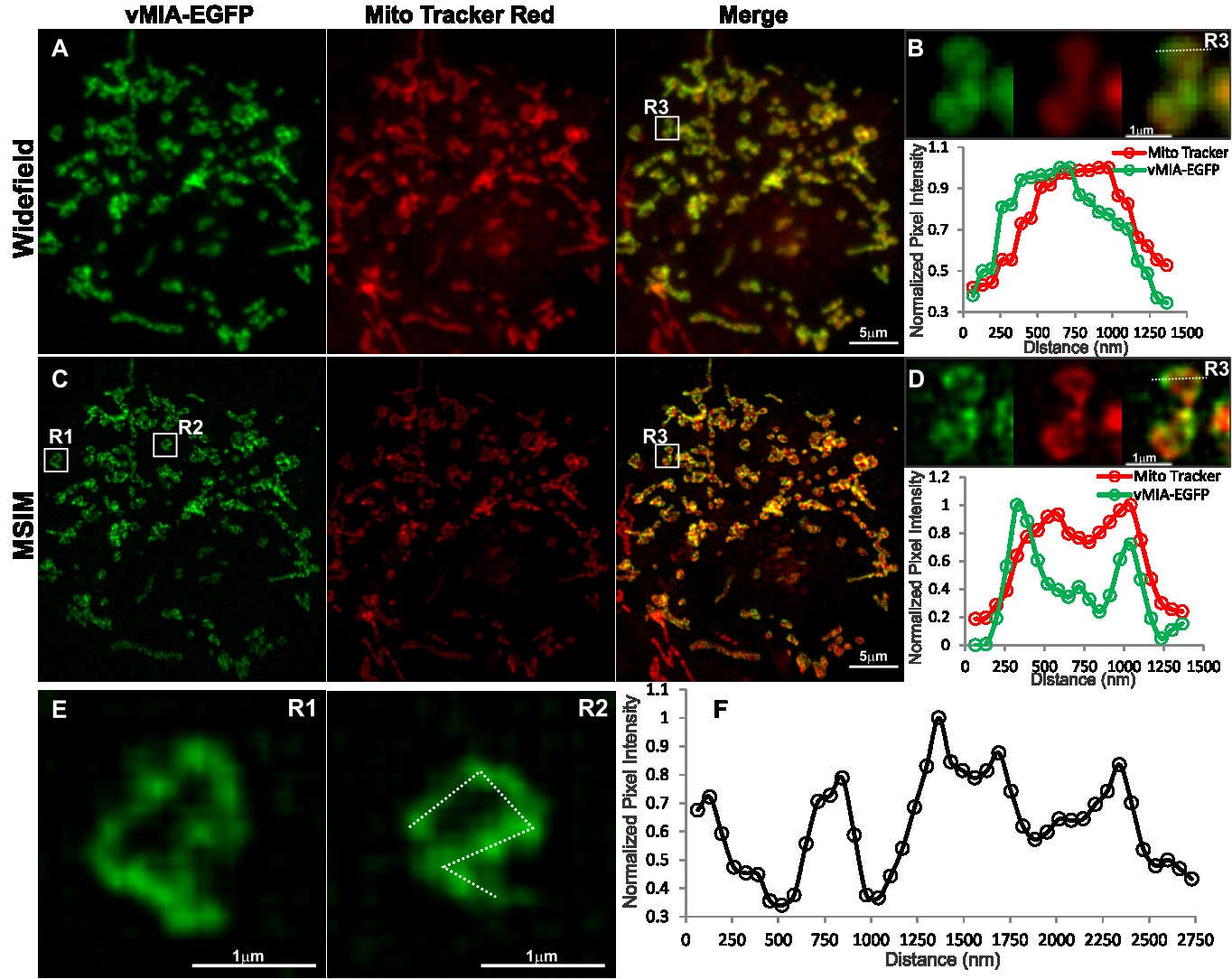
2.3. MSIM Imaging of vMIA and Mitochondrial Marker
With a restricted set of compatible fluorophores that can be used for multicolor superresolution
imaging by GSTED, for simultaneous superresolution imaging of vMIA-EGFP in context of organelle
Viruses 2014, 6
markers we made use of an alternative superresolution imaging approach—MSIM. We performed
MSIM imaging of doxycycline (Dox) treated HFFs dually transfected with the tetracycline
transactivator [53] and the TRE-Tight promoter driving vMIA-EGFP (Figure 7). We observed that
vMIA localized distinctly from the mitochondrial marker we used here—MitoTracker Red, which
appears to localize to the IMS [38]. As expected, widefield imaging of vMIA-EGFP and MitoTracker
Red showed predominant colocalization of the two fluorophores (Figure 7A). With the observed
spatial resolution of 302 nm for this image there was little resolution of the vMIA and MitoTracker
labeling (Figure 7B). The overlap of the signals was documented by the intensity profile of each
fluorophore along the pixels marked by the dotted line shown in the zoom of the region R3
(Figure 7B). MSIM images of the two fluorophores for this cell showed improved resolution of
vMIA-EGFP and MitoTracker Red (Figure 7C). Intensity profile of MSIM image of each fluorophore
along the pixels marked by the dotted line shown in the zoom of the region R3 showed partial
separation of the vMIA-EGFP fluorescence peaks from those of the MitoTracker fluorescence peaks
such that the bimodal vMIA peaks are farther away to the outside of the bimodal MitoTracker peaks
(FWHM = 120nm) (Figure 7D). This distribution of the two markers is in agreement with the vMIA
localization to the OMM, surrounding the IMS localized MitoTracker Red. Furthermore, MSIM
imaging in individual mitochondrion (R1 and R2 in Figure 7E) confirmed our previous observations of
vMIA localization in clusters at the OMM. The presence of vMIA clusters was verified by intensity
profile of MSIM image of each fluorophore along the pixels marked by the dotted line drawn along the
rim of a mitochondrion in R2 (Figure 7F). These vMIA clusters were obscured by diffraction in
widefield imaging (Figure 7B) and confocal microscopy (Figures 2–4) but detected by superresolution
GSTED imaging (Figures 5 and 6). Together, these results show that MSIM partially resolves vMIA
location at mitochondria periphery, away from IMS, and in clusters of diffraction limited size. The ER
and OMM make contacts at the MAM. In addition, using superresolution microscopy multiple
mitochondrial proteins including the OMM VDAC1, VDAC3 and Tom 20 proteins have been shown
to exist in clusters as functional mitochondrial complexes [46,47,54]. vMIA clustering could represent
the contact sites between the ER and OMM or could indicate the vMIA association with functional
complexes at the OMM. Use of above approaches allowed us to narrow down the size of vMIA
clusters to be in the range of 100 nm, but this is close to the resolution of imaging modalities used.
2.4. PALM Imaging of PA-mCherry-vMIA
To visualize vMIA clusters by yet another independent superresolution approach and obtain a better
estimate of the size of vMIA clusters, we made use of the pointillistic imaging superresolution imaging approach PALM. Here, we expressed vMIA tagged with PAmCherry, a photoactivatable red fluorescent protein [55], in HFFs and visualized them using 2D PALM imaging (Figure 8). Most of the molecules in this image localized with precision better than 25nm, but here we have conservatively rendered all molecules which have been localized to <25 nm precision as 2D Gaussian distributions with 25 nm sigmas. Similar to our MSIM and GSTED results above, qualitative observations suggest a non-uniform distribution of vMIA-PAmCherry on the periphery of the mitochondria when rendered in this manner. PALM imaging also affords a second, rather straightforward analysis of molecule distributions, which is relevant to this work, cluster analysis. Several methods are developed for this
Viruses 2014, 6
sort of analysis, but we opted for pair correlation analysis in quantitatively determining which regions
of each mitochondrion displayed increased vMIA protein densities.
Figure 7. Imaging vMIA localization using widefield microscopy and MSIM. HFFs were
transfected with vectors expressing TRE-tight promoter-vMIA-EGFP (green) and its
tetracycline controlled transactivator (tTA) [53]. Transfected cells were Dox-treated
(0.2 µg/mL) for one hour, MitoTracker Red treated (0.5 µM for 5 min, red) and methanol
fixed as described [23]. 25 slices (0.2 µm step size) were collected using 488 nm and 561
nm excitation lasers. For each channel, 256 multifocal excited images were collected as the
excitation array was stepped 1 pixel in a 16 × 16 grid pattern. (A) Summation of the
images produces the widefield fluorescence image, which shows high degree of
co-localization between vMIA-EGFP and MitoTracker Red. (B) Zoomed images of
mitochondria in the region marked R3 are shown and the plot below shows the intensity
profile through the region marked by dotted line. (C) The MSIM image is shown. (D) The
zoomed images of region R3 and the line scan for the region marked by dotted line show
the improved ability to resolve the vMIA-EGFP and MitoTracker Red staining through the
use of MSIM. (E) MSIM images of two mitochondria (R1 and R2) are shown. (F) Intensity
profile through the perimeter of mitochondrion in R2 marked by dotted line is shown in the
plot. This indicates non-uniform distribution of vMIA-GFP along the mitochondrial
membrane. For higher resolution images, see Supplemental Figure S6.
Viruses 2014, 6
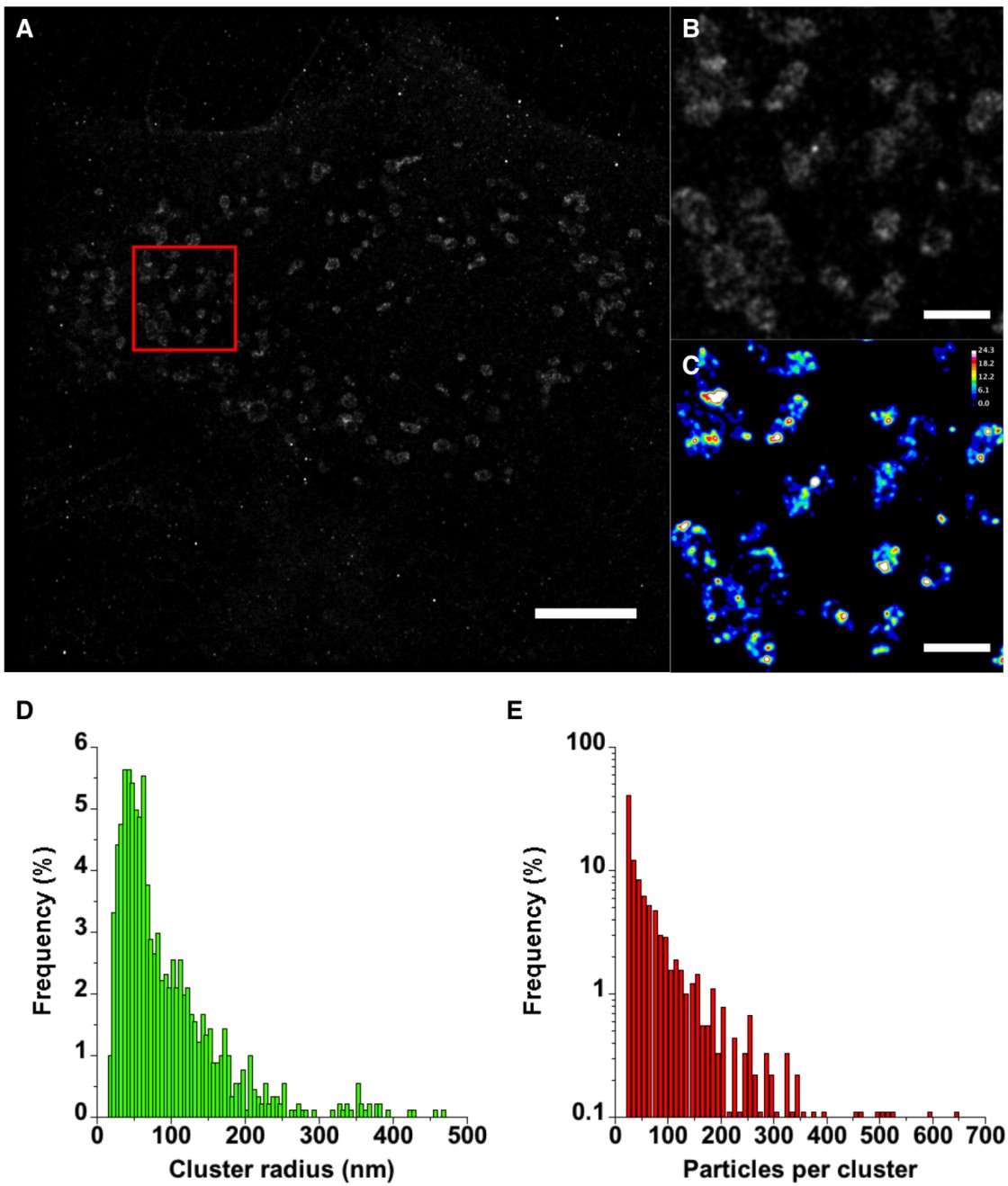
Figure 8. PALM imaging of vMIA-PAmCherry1. HFFs were transfected with
vMIA-PAmCherry and imaged by PALM. (A) The image shows 301666 molecules that
were localized to ≤25 nm precision out of 515207. Molecules are plotted as
two-dimensional Gaussian distributions with 25 nm standard deviations. (B) The image
shows a small region in (A) indicated by the red square. The molecule positions in a binary
image were used in pair correlation analysis and the maximum g(r) derived from using
each molecule as a reference point was plotted as the pixel values. (C) The image shows
the same region in the red square color-coded to display the varying degrees of molecule
densities. The Look Up Table (LUT) and calibration bar are included in the upper right
corner. The distributions of cluster sizes (D) and the number of particles per cluster (E)
were determined from the pair correlation analysis results. The maximum cluster radius
was limited to 500 nm and the minimum number of particles per cluster was limited to 20.
Please note that the Y axis in (E) is a log scale to better indicate the lower percentages at
the higher particle numbers. The scale bar in (A) is 5 μm and in (B, C) is 1 μm. High
resolution images of the entire field of view are available in the SOM (see Supplemental
Figures S7, S10 and S11).
Viruses 2014, 6
In these analyses, the function g(r) is determined by comparing the protein density in a local ―shell‖
region around each position with the average density of all molecules of interest (see Supplemental Figure S8). Simulated images containing various levels of clustering are shown in Supplemental Figure S9A–D. For these, the factor g is plotted as a function of the radius and for images containing no clusters, this remains close to 1 as the radius decreases toward zero (Supplemental Figure S9E). However, in images with clusters, the g(r) deviates from 1 at approximately the diameter of the clusters (Supplemental Figure S9E). If the cluster radii increase, the g(r) deviates at a higher value of r. If the molecule density in the cluster increases, the maximum g(r) increases. Thus, the maximum g(r) can reflect the relative amplitude of the molecule density surrounding each reference particle. This is important for our simple analysis in which we compare the local protein density with the whole image density. With these criteria, the entire mitochondrion could be considered a cluster. In our analyses, we used the average density of all molecules in the image localized to <25 nm precision. We plotted the g(r) maximum value obtained for each reference molecule in a new image at the appropriate location (Supplemental Figure S10). Indeed, we find that most of the molecules localized to the mitochondria show g(r) values greater than 1, but we also note that regions resembling the clusters observed with MSIM and GSTED have much higher g(r) values than surrounding peripheral regions (Figure 8). Using the results from pair correlation analysis, the cluster sizes (Figure 8D) and the number of particles per cluster (Figure 8E) were determined. Cluster radii show a broad distribution with a mean radius of approximately 95 nm and suggest that a majority are <100 nm. The number of particles per cluster is also broadly distributed and indicates that a majority of the clusters (approximately 60%) contain 50 particles or less.
3. Experimental Section
3.1. Cell Culture and Lipofection
HFFs were cultured in Dulbecco's Modified Eagle's medium containing 10% fetal calf serum (FCS,
Hyclone, Logan, UT), 100 U/mL of penicillin, 100 μg/mL of streptomycin, 2 mM L-Glutamine (Life Technologies, Grand Island, NY) as previously described [56]. U373-Tet-ON cells were also cultured in the 10% FCS, 100 U/mL of penicillin, 100 μg/mL of streptomycin, 2 mM L-Glutamine but maintained under selective pressure using 0.6 mg/mL Geneticin (Life Technologies).
Cells were seeded at 20%–50% confluency on sterile 18 mm (for confocal or GSTED) or 25 mm
(for PALM and MSIM) cover slips in six-well plates (9.72 cm2 per well). Twenty-four hours later cells were transiently transfected using Lipofectamine 2000 (Life Technologies) suspended in Opti-MEM (Life Technologies), according to the manufacturer's protocols. DNA (µg):lipid (µL) ratios for transfection were at 1:1.77, with approximately 0.5 µg total DNA used per cm2 of available plating surface area. Cells were harvested 22-25 hours after transfection by fixation with methanol or 4% PFA in PBS and mounted with Prolong Gold Antifade. GSTED imaging was performed on live U373-Tet-ON cells treated with Dox (0.5 µM).
Viruses 2014, 6
3.2. Construction of pTRE-Tight Promoter-vMIA-EGFP
The vMIA/UL37x1 protein-EGFP open reading was isolated from p1242 [15] by restriction
enzyme digestion with EcoRI and NotI. The fragment was ligated into EcoRI/NotI digested TRE-Tight vector (Clontech, Mountain View, CA, USA) and the ligation product was transformed into competent E. coli (strain DH5 alpha).
3.3. Confocal Microscopy
The confocal images were acquired using the Olympus FV1000 confocal microscope. An
UPlanSApo100x/1.40NA oil objective was used to obtain an oversampled 1024 × 1024 image with 49 nm pixel and a z-stack with a step size of 120 nm. The Mito-BFP and the vMIA-EGFP were sequentially excited using a 405nm diode laser and a 488nm Argon laser, respectively, and collected between 425 nm–475 nm and 500 nm–545 nm in the spectral detectors. In the triple labeled cells, ss-RFP-KDEL was also sequentially collected using 575 nm–675 nm filter after excitation with a 559 nm diode laser.
3.4. GSTED Microscopy
The GSTED images were acquired using a Leica TCS SP5 gated STED (GSTED) microscope that
was equipped with a super continuum white light laser (WLL) and hybrid detectors adapted for time gated imaging which allow elimination of low spatial frequency from the final super resolved image [57]. The 592 nm depletion laser delivers 0.3W at the focal plane. A HCX PL APO 100× 1.40 NA oil objective was used to obtain a 1600 × 1600 image with a 24.2 nm pixel. The DsRed-Mito confocal image was acquired using the 560 nm excitation and collected between 569 nm–665 nm. A sequential GSTED image of UL37-EGFP was obtained using a 488 nm excitation, 592 nm depletion, time gating from 1–8 ns, and collected between 500 nm–544 nm. Using the tunability of the white light laser and prism based spectral emission. ―Lambda lambda‖ scans were acquired for an excitation range of 470 nm–590 nm and an emission range of 485 nm–605 nm. Both excitation and emission scans were taken with 10 nm wavelength steps between measurements and was used to confirm the excitation and emission properties of the EGFP fluorophore imaged on this microscope in the GSTED or confocal mode.
3.5. Deconvolution Analysis
Blurring due to out of focus signal and Poisson noise in the confocal and GSTED images was
removed by carrying out deconvolution using the Huygens Essential software supplied by Scientific Volume Imaging B.V. (Hilversum, The Netherlands). For confocal images, deconvolution was done using 3D images acquired as described above. For GSTED images, 2D deconvolution was done on the using images acquired as discussed below. For generating pixel intensity plots images were imported and analyzed in the Metamorph Premier (7.7.0) software supplied by Molecular Devices, LLC (Sunnyvale, CA, USA).
Viruses 2014, 6
3.6. MSIM
The MSIM microscope used for these experiments is a homebuilt machine using an Olympus IX-71
widefield microscope as previously described [58] and with modifications detailed below. A 100 mW 405 nm Cube laser (Coherent, Inc., Santa Clara, CA, USA), 50 mW 488 laser (Oxxius, Lannion, France, LBX-488-50-CIR-PP), 50 mW 561 Sapphire laser (Coherent, Inc.), and 100 mW 640 nm Cube laser (Coherent, Inc.) served as illumination sources for blue, green, red, and far-red fluorophores, respectively. The light from each laser was filtered by placing a 405/10 nm BrightLine single-band bandpass filter (Semrock Inc., Rochester, NY, USA, FF01-405-10/25) at the aperture of the 405 laser, a 488/6 nm BrightLine single-band bandpass filter (Semrock Inc., FF01-488-6/25) at the aperture of the 488 laser, a 561/4 nm BrightLine single-band bandpass filter (Semrock Inc., FF01-561-6/25) at the aperture of the 561 laser, and a 640/8 nm MaxDiode laser clean-up filter (Semrock Inc., LL01-640-8/12.5) at the aperture of the 640 laser. The laser beams were collimated or expanded and collimated with insertion of pairs of lenses after the clean-up filters. The 405 laser has a 2× telescope consisting of a 50 mm focal length lens (Thorlabs Inc., Newton, NJ, USA, LA1131-A) and a 100 mm focal length lens (Thorlabs Inc., LA1509-A). The 488 laser has a 1× telescope consisting of a pair of 100 mm focal length lenses (Thorlabs Inc., LA1509-A). The 561 laser has a 3× telescope consisting of a 50 mm focal length lens (Thorlabs Inc., LA1131-A) and a 150 mm focal length lens (Thorlabs Inc., LA1433-A). The 640 laser has a 2× telescope consisting of a 50 mm focal length lens (Thorlabs Inc., LA1131-A) and a 100 mm focal length lens (Thorlabs Inc., AC254-100-A). Mechanical shutters along with acousto-optic tunable filter (AA Opto-electronic Inc., Orsay, France, AOTFnC-400.650) controlled by Micro-Manager [59] allowed for laser wavelength selection, laser power tuning and laser shuttering. The laser beams were expanded 5× using a 40 mm focal length lens (Thorlabs Inc., AC254-40-A) and a 200 mm focal length lens (Thorlabs Inc., AC254-200-A). Collected fluorescence was filtered through a 480/40 bandpass filter (Chroma Technology Corp, Bellows Falls, VT, USA D480/40m) for 405 nm excitation, a 525/45 bandpass filter (Semrock, FF01-525/45-25) for 488 nm excitation, a 600/37 bandpass filter (Semrock, FF01-600/37-25) for 561 nm excitation, and a EdgeBasic 635 longpass filter (Semrock, BLP01-635R-25) for 640 nm excitation. All other components in the excitation and emission paths, such as optical components, 2D galvanometer, z-stage and camera, are the same as previously published [58].
3.7. MSIM Data Collection and Analysis
MSIM data were collected for each slice as previously published [58] except the galvanometer was
stepped in a 16 × 16 grid to collect 256 frames for each 512 × 512 pixel wide field of view. Post-processing was performed as previously published [58] on freely available software [60]. Deconvolution was performed on the MPSS images using a program written in Python (freely available at [60] implementing Richardson–Lucy deconvolution [58,61–63].
3.8. PALM Data Analysis
Single molecule images were analyzed as previously described [64] using PeakSelector (a program
written in IDL by Harald Hess and Gleb Shtengel, Howard Hughes Medical Institute, Janelia Farms,
Viruses 2014, 6
Ashburn, VA, USA). The molecule localization data were output as an Ascii file which was used by an
ImageJ macro (available upon request) [65] to plot the molecule positions and precisions on images
with 5 nm pixels.
Pair correlation analysis was performed on binary images displaying peak positions rendered as
single 5 nm pixels. The g(r) function was determined for each particle by using the equation
g(r) = ρ(δr)/ρ(AN)
where each particle is treated as a reference particle. The particle density in a ―shell‖ region of δr is determined and then divided by the particle density in a region containing all of the particles of interest, AN. In these analyses, the entire image was used as AN (Supplemental Figure S8). The analysis was performed using an ImageJ macro (available upon request) run in FIJI [66]. Briefly, the particle density ρ in region AN, ρ(δr), was determined by dividing the total number of rendered molecules by the area of the image. The particle density ρ in region δr, ρ(δr), was determined by first counting the number of particles in areas r1 and r2 (Supplemental Figure S8). The number of particles in region δr was determined by subtracting the r1 count from the r2 count. The area of δr was determined using πr2 by subtracting the area the r1 region from the area of the r2 region. The ρ(δr) was calculated by dividing the δr count by the area of δr.
The g(r) as a function of the radius r1 for each peak was fit with an exponential equation
g(r) = A × exp(–(1/B) × r) + C
where A is the amplitude, B is the radius of the cluster, and C is the offset. Only peaks with g(r) fitted with a correlation coefficient R2 > 0.9 were rendered in the g(r) image. To display these data, the molecule peak position image was rendered by setting the pixel values for each particle to the maximum g(r) determined for that peak when using it as a reference in the analysis (Supplemental Figure S10). A color coded LUT with calibration is included for comparison of different regions of the cell. Additional processing on this image included a Gaussian blur with a 25 nm sigma to represent the uncertainty in molecule localization (Supplemental Figure S11).
4. Conclusions
Because superresolution microscopy is an emerging technology, it has had very limited use in the
study of viruses. To date, superresolution imaging has primarily focused on RNA viruses including imaging of rotavirus virions [67], influenza hemagglutinin clusters [68], HIV assembly [64,69,70] and HIV transfer [71]. More recently, single copy viral genomes including those of DNA viruses have been imaged using superresolution microscopy [72]. To our knowledge, our studies are the first superresolution imaging of any human herpesvirus protein, in particular of a HCMV vMIA.
HCMV vMIA imaging by confocal/deconvolution microscopy indicated its OMM localization,
which was confirmed by GSTED, MSIM, and PALM imaging. Additionally these imaging modalities all demonstrate that at the OMM vMIA is present in clusters, indicating nanoscale localization of HCMV vMIA at the mitochondrial periphery, away from IMS and matrix. While this clustering is also suggested by deconvolved images obtained by the confocal microscopy, diffraction limits the ability to visualize the distribution of MAM clusters. This indicates that the vMIA domains are generally spaced closer than the diffraction limit. This inference has been supported by the ability to resolve them by
Viruses 2014, 6
each of the superresolution imaging approaches used here. Use of PALM imaging allowed us to
estimate the sizes of these clusters at approximately 95 nm, which is in agreement with the cluster
sizes indicated by the GSTED and MSIM approaches.
Intriguingly, clusters of cellular mitochondrial proteins have been observed using superresolution
microscopy. STED microscopy found that MINOS, which maintains IMM morphology, forms clusters often in an ordered inner mitochondrial distribution [45]. Tom 20 and Tom22, components of the translocase of the outer mitochondrial membrane, are clustered at the OMM with densities that correlate with mitochondrial membrane potential [73]. VDAC1 and VDAC3 have also been found by superresolution imaging to be distributed in clusters in the OMM. Cytosolic hexokinase I which associates with VDAC3 is partly also localized in clusters with VDAC3 [46,47]. Our superresolution imaging suggests that vMIA may target and associate with clustered OMM proteins. Alternatively, vMIA traffics efficiently to the MAM [20–23,26,74]. vMIA clustering may represent sites of contacts between the ER and OMM, where vMIA could be transferred between the two organelles. The use of vMIA mutants and known cellular markers of MAM should allow us to distinguish these possibilities.
Superresolution imaging has the clear potential to provide valuable insight into the nanoscale
organization of viral machineries, which provide essential replicative processes. Case in point is the detection of non-uniform distribution of vMIA at the OMM. This distribution was not detectable by conventional microscopy because of its diffraction limited resolution. Further, this technology allows virologists to study how viruses alter cellular organelles to establish replication compartments and virus assembly. However, the superresolution approach to be used should be carefully selected. Our approach here involved three techniques with complementary advantages in nanoscale studies of vMIA localization. GSTED relies primarily on imaging of one fluorophore impacting on the ability to place the viral proteins in the in situ biological context of cellular organelles or structures. Secondary markers can be imaged by GSTED using the appropriately labeled secondary antibodies (e.g., Alexa 532). GSTED provides greater resolution (<50 nm) than MSIM. However, use of primary and secondary antibodies can adversely affect precision of protein localization at the nanoscale resolution. Nonetheless, because of its resolution, GSTED provided compelling evidence for vMIA clustering in mitochondria and localization at the OMM. Conversely, MSIM is lower resolution than GSTED microscopy. However, MSIM makes use of the same fluorophores used for conventional confocal microscopy. This enables imaging of viral proteins in the context of multiple fluorophore tagged proteins from different sub-cellular compartments. For our experiments, MSIM provided sufficient resolution to confirm the location and clustering of vMIA at the OMM.
PALM imaging provides the highest resolution used in these studies but imaging sufficient
molecules to obtain meaningful information requires hours of acquisition and post-acquisition processing. This limits most PALM imaging to fixed specimens. Given the increasing palette of photoactivatable fluorophores and increasing availability of these powerful microscopy techniques much can be learned by their application to multiple viruses and their processes.
Here, we have used three different superresolution techniques with multiple markers to image the
localization and clustering of vMIA. These all confirm the presence of the clusters on the outer membranes of mitochondria and are helping to define the cluster physical characteristics. In general, the clusters are found closer together than the limits of resolution by widefield and confocal microscopy. Examples of these include the plot profiles in Figures 5–7 compared with the widefield or
Viruses 2014, 6
confocal counterparts. These results show that given time, the imaging techniques used here will help
discern on a sub-mitochondria scale where are the components of MAMs located in relation to each
other as well as other cellular players.
A second important finding here is that we may put a conservative upper limit on the
diffraction-limited vMIA cluster size of approximately 150 nm. Since this comes from the data collected with three independent imaging techniques, it provides a more reasonable estimate than simply relying on a single technique.
Why is the size of importance? At these scales, small cluster sizes dictate the maximum number of
molecules which can fit inside. Given the number of processes in which MAMs are known to play a role, this imposes limits on the number of molecules which can participate in signaling events. Thus, every MAM may not have all of the components to initiate all processes and this brings up another nagging question in the MAM field. Are the MAMs homogeneous or have differing compositions which imply different functions? Now that these structures can be readily imaged with optical techniques using multiple color markers, answers to this question will be far more straightforward to derive than with previous efforts. In summary, our studies show that superresolution imaging provides valuable insight into sub-diffraction resolution of viral protein location, particularly in the sub-mitochondrial compartments, and into their clustered arrangement.
This work was partially funded by the NSF grant (MCB 1244509) to ACP and JKJ. The confocal
microscopy imaging was supported by a core grant (1P30HD40677) to the Children's Intellectual and Developmental Disabilities Research Center. This work was also supported by the Intramural Research Program of the National Institutes of Health including the National Institute of Biomedical Imaging and Bioengineering.
The authors thank Yan Su for construction of pTRE-Tight promoter-vMIA-EGFP plasmid, Jennifer
Lippincott-Schwartz for providing ss-RFP-KDEL and Gary Thomas for the gift of U373-Tet-ON cells. We thank Maria Ingaramo for technical assistance with the MSIM image collection and analysis. We thank Andrew York and Hari Shroff for sharing MSIM analysis and deconvolution software. We thank Harald Hess for the use of the PALM analysis software, PeakSelector.
Author Contributions
S.B. and S.C.S. performed confocal microscopy, K.S., M.L., and V.P. performed transfections,
E.W. and K.R. collected and analyzed the MSIM data, J.B. performed GSTED imaging, S.B., S.C.S. and J.K.J. performed the deconvolution and analysis of the confocal and GSTED images, G.H.P. collected and analyzed the PALM images, G.H.P., J.K.J. and A.C.P. designed experiments, analyzed results and wrote the manuscript. All edited the manuscript.
Conflicts of Interest
Jonathan Boyd is an employee of Leica Microsystems. The other authors declare no conflict
Viruses 2014, 6
References and Notes
1. Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends
Cell Biol. 2009, 19, 81–88.
2. Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; de Marchi, E.;
Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (MAMs) as
hotspot Ca(2+) signaling units. Adv. Exp. Med. Biol. 2012, 740, 411–437.
3. Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One
touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469.
4. Szabadkai, G.; Bianchi, K.; Varnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.;
Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911.
5. Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.;
Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor
Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283.
6. Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.;
Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes
mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011,
124, 2143–2152.
7. Castanier, C.; Arnoult, D. Mitochondrial localization of viral proteins as a means to subvert host
defense. Biochim. Biophys. Acta 2010, 1813, 575–583.
8. Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can
permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634.
9. Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-
associated membranes. J. Biol. Chem. 2000, 275, 34534–34540.
10. Hayashi, T.; Fujimoto, M. Detergent-resistant microdomains determine the localization of
sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 2010,
77, 517–528.
11. Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Lipid raft connection between extrinsic and
intrinsic apoptotic pathways. Biochem. Biophys. Res. Commun. 2009, 380, 780–784.
12. Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the
RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138.
13. Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated
endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by
hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595.
14. Reboredo, M.; Greaves, R.F.; Hahn, G. Human cytomegalovirus proteins encoded by UL37 exon
1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus
replication. J. Gen. Virol. 2004, 85, 3555–3567.
15. Sharon-Friling, R.; Goodhouse, J.; Colberg-Poley, A.M.; Shenk, T. Human cytomegalovirus
pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA
2006, 103, 19117–19122.
Viruses 2014, 6
16. Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.;
Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded
mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad.
Sci. USA 1999, 96, 12536–12541.
17. Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.;
Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not
Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci.
USA 2004, 101, 7988–7993.
18. Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.;
Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits
Bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614.
19. Hayajneh, W.A.; Colberg-Poley, A.M.; Skaletskaya, A.; Bartle, L.M.; Lesperance, M.M.;
Contopoulos-Ioannidis, D.G.; Kedersha, N.L.; Goldmacher, V.S. The sequence and antiapoptotic
functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are
conserved in multiple primary strains. Virology 2001, 279, 233–240.
20. Mavinakere, M.S.; Williamson, C.D.; Goldmacher, V.S.; Colberg-Poley, A.M. Processing of
human cytomegalovirus UL37 mutant glycoproteins in the endoplasmic reticulum lumen prior to
mitochondrial importation. J. Virol. 2006, 80, 6771–6783.
21. Bozidis, P.; Williamson, C.D.; Colberg-Poley, A.M. Mitochondrial and secretory human
cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells.
J. Virol. 2008, 82, 2715–2726.
22. Bozidis, P.; Williamson, C.D.; Wong, D.S.; Colberg-Poley, A.M. Trafficking of UL37 proteins
into mitochondrion-associated membranes during permissive human cytomegalovirus infection.
J. Virol. 2010, 84, 7898–7903.
23. Williamson, C.D.; Colberg-Poley, A.M. Intracellular Sorting Signals for Sequential Trafficking of
Human Cytomegalovirus UL37 Proteins to the Endoplasmic Reticulum and Mitochondria.
J. Virol. 2010, 84, 6400–6409.
24. Colberg-Poley, A.M.; Patel, M.B.; Erezo, D.P.; Slater, J.E. Human cytomegalovirus UL37
immediate-early regulatory proteins traffic through the secretory apparatus and to mitochondria.
J. Gen. Virol. 2000, 81, 1779–1789.
25. Williamson, C.D.; Colberg-Poley, A.M. Access of viral proteins to mitochondria via
mitochondria-associated membranes. Rev. Med. Virol. 2009, 19, 147–164.
26. Williamson, C.D.; Zhang, A.; Colberg-Poley, A.M. The human cytomegalovirus protein UL37
exon 1 associates with internal lipid rafts. J. Virol. 2011, 85, 2100–2111.
27. Zhang, A.; Hildreth, R.L.; Colberg-Poley, A.M. Human cytomegalovirus inhibits apoptosis by
proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts.
J. Virol. 2013, 87, 5657–5668.
28. Seo, J.Y.; Yaneva, R.; Hinson, E.R.; Cresswell, P. Human cytomegalovirus directly induces the
antiviral protein viperin to enhance infectivity. Science 2011, 332, 1093–1097.
29. Seo, J.Y.; Cresswell, P. Viperin regulates cellular lipid metabolism during human
cytomegalovirus infection. PLoS Pathog. 2013, 9, e1003497.
Viruses 2014, 6
30. Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial
morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243.
31. Ma, J.; Edlich, F.; Bermejo, G.A.; Norris, K.L.; Youle, R.J.; Tjandra, N. Structural mechanism
of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. USA 2012, 109,
20901–20906.
32. Poncet, D.; Pauleau, A.L.; Szabadkai, G.; Vozza, A.; Scholz, S.R.; Le Bras, M.; Briere, J.J.;
Jalil, A.; Le Moigne, R.; Brenner, C.; et al. Cytopathic effects of the cytomegalovirus-encoded
apoptosis inhibitory protein vMIA. J. Cell Biol. 2006, 174, 985–996.
33. McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of mitochondrial networks
by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of
apoptosis. J. Virol. 2003, 77, 631–641.
34. Roumier, T.; Szabadkai, G.; Simoni, A.M.; Perfettini, J.L.; Paulau, A.L.; Castedo, M.;
Metivier, D.; Badley, A.; Rizzuto, R.; Kroemer, G. HIV-1 protease inhibitors and
cytomegalovirus vMIA induce mitochondrial fragmentation without triggering apoptosis. Cell
Death Differ. 2006, 13, 348–351.
35. McCormick, A.L.; Roback, L.; Mocarski, E.S. HtrA2/Omi terminates cytomegalovirus infection
and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog. 2008,
4, e1000063.
36. Altan-Bonnet, N.; Sougrat, R.; Liu, W.; Snapp, E.L.; Ward, T.; Lippincott-Schwartz, J. Golgi
inheritance in mammalian cells is mediated through endoplasmic reticulum export activities.
Mol. Biol. Cell 2006, 17, 990–1005.
37. Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules
mark sites of mitochondrial division. Science 2011, 334, 358–362.
38. Shim, S.H.; Xia, C.; Zhong, G.; Babcock, H.P.; Vaughan, J.C.; Huang, B.; Wang, X.; Xu, C.;
Bi, G.Q.; Zhuang, X. Super-resolution fluorescence imaging of organelles in live cells with
photoswitchable membrane probes. Proc. Natl. Acad. Sci. USA 2012, 109, 13978–13983.
39. Shao, L.; Kner, P.; Rego, E.H.; Gustafsson, M.G. Super-resolution 3D microscopy of live whole
cells using structured illumination. Nat. Methods 2011, 8, 1044–1046.
40. Arnoult, D.; Skaletskaya, A.; Estaquier, J.; Dufour, C.; Goldmacher, V.S. The murine
cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial
outer membrane permeabilization. Apoptosis 2008, 13, 1100–1110.
41. Biggs, D.S. 3D deconvolution microscopy. Curr. Protoc. Cytom. 2010, doi:10.1002/0471142956.
42. York, A.G.; Parekh, S.H.; Dalle Nogare, D.; Fischer, R.S.; Temprine, K.; Mione, M.; Chitnis,
A.B.; Combs, C.A.; Shroff, H. Resolution doubling in live, multicellular organisms via multifocal
structured illumination microscopy. Nat. Methods 2012, 9, 749–754.
43. Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158.
44. Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission:
stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782.
45. Jans, D.C.; Wurm, C.A.; Riedel, D.; Wenzel, D.; Stagge, F.; Deckers, M.; Rehling, P.; Jakobs, S.
STED super-resolution microscopy reveals an array of MINOS clusters along human
mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 8936–8941.
Viruses 2014, 6
46. Neumann, D.; Buckers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-color STED microscopy
reveals different degrees of colocalization between hexokinase-I and the three human VDAC
isoforms. PMC Biophys. 2010, 3, 4.
47. Singh, H.; Lu, R.; Rodriguez, P.F.; Wu, Y.; Bopassa, J.C.; Stefani, E.; Toro, L. Visualization and
quantification of cardiac mitochondrial protein clusters with STED microscopy. Mitochondrion
2012, 12, 230–236.
48. Lippincott-Schwartz, J.; Patterson, G.H. Photoactivatable fluorescent proteins for diffraction-
limited and super-resolution imaging. Trends Cell Biol. 2009, 19, 555–565.
49. Pauleau, A.L.; Larochette, N.; Giordanetto, F.; Scholz, S.R.; Poncet, D.; Zamzami, N.;
Goldmacher, V.S.; Kroemer, G. Structure-function analysis of the interaction between Bax and
the cytomegalovirus-encoded protein vMIA. Oncogene 2007, 26, 7067–7080.
50. Hildreth, R.L.; Bullough, M.D.; Zhang, A.; Chen, H.L.; Schwartz, P.H.; Panchision, D.M.;
Colberg-Poley, A.M. Viral mitochondria-localized inhibitor of apoptosis (UL37 exon 1 protein)
does not protect human neural precursor cells from human cytomegalovirus-induced cell death.
J. Gen. Virol. 2012, 93, 2436–2446.
51. Crump, C.M.; Hung, C.H.; Thomas, L.; Wan, L.; Thomas, G. Role of PACS-1 in trafficking of
human cytomegalovirus glycoprotein B and virus production. J. Virol. 2003, 77, 11105–11113.
52. Colberg-Poley, A.M.; Williamson, C.D. Intracellular sorting and trafficking of cytomegalovirus
proteins during permissive infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention, 2nd ed.; Reddehase, M.J., Ed.; Caister Academic Press/Horizon: Norwich, UK, 2013; Volume I, pp. 196–229.
53. Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by
tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551.
54. Watanabe, S.; Punge, A.; Hollopeter, G.; Willig, K.I.; Hobson, R.J.; Davis, M.W.; Hell, S.W.;
Jorgensen, E.M. Protein localization in electron micrographs using fluorescence nanoscopy.
Nat. Methods 2011, 8, 80–84.
55. Subach, F.V.; Patterson, G.H.; Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J.;
Verkhusha, V.V. Photoactivatable mCherry for high-resolution two-color fluorescence
microscopy. Nat. Methods 2009, 6, 153–159.
56. Adair, R.; Liebisch, G.W.; Su, Y.; Colberg-Poley, A.M. Alteration of cellular RNA splicing and
polyadenylation machineries during productive human cytomegalovirus infection. J. Gen. Virol.
2004, 85, 3541–3553.
57. Vicidomini, G.; Moneron, G.; Han, K.Y.; Westphal, V.; Ta, H.; Reuss, M.; Engelhardt, J.;
Eggeling, C.; Hell, S.W. Sharper low-power STED nanoscopy by time gating. Nat. Methods 2011,
8, 571–573.
58. Ingaramo, M.; York, A.G.; Wawrzusin, P.; Milberg, O.; Hong, A.; Weigert, R.; Shroff, H.;
Patterson, G.H. Two-photon excitation improves multifocal structured illumination in thick
scattering tissue. Proc. Natl. Acad. Sci. USA 2014, in press.
59. Edelstein, A.; Amodaj, N.; Hoover, K.; Vale, R.; Stuurman, N. Computer control of microscopes
using microManager. Curr. Protoc. Mol. Biol. 2010, doi:10.1002/0471142727.mb1420s92.
60. York, A.G. MSIM Superresolution Fluorescence Microscopy of Multicellular Organisms.
Available online: http://code.google.com/p/msim/ (accessed on 3 May 2012).
Viruses 2014, 6
61. Richardson, W.H. Bayesian-based iterative method of image restoration. JOSA 1972, 62, 55–59.
62. Lucy, L. An iterative technique for the rectification of observed distributions. Astron. J. 1974, 79,
63. York, A.G.; Chandris, P.; Nogare, D.D.; Head, J.; Wawrzusin, P.; Fischer, R.S.; Chitnis, A.;
Shroff, H. Instant super-resolution imaging in live cells and embryos via analog image processing.
Nat. Methods 2013, 10, 1122–1126.
64. Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.;
Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at
nanometer resolution. Science 2006, 313, 1642–1645.
65. Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis.
Nat. Methods 2012, 9, 671–675.
66. Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.;
Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image
analysis. Nat. Methods 2012, 9, 676–682.
67. Willig, K.I.; Kellner, R.R.; Medda, R.; Hein, B.; Jakobs, S.; Hell, S.W. Nanoscale resolution in
GFP-based microscopy. Nat. Methods 2006, 3, 721–723.
68. Gudheti, M.V.; Curthoys, N.M.; Gould, T.J.; Kim, D.; Gunewardene, M.S.; Gabor, K.A.; Gosse,
J.A.; Kim, C.H.; Zimmerberg, J.; Hess, S.T. Actin mediates the nanoscale membrane organization
of the clustered membrane protein influenza hemagglutinin. Biophys. J. 2013, 104, 2182–2192.
69. Roy, N.H.; Chan, J.; Lambele, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral
assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525.
70. Grover, J.R.; Llewellyn, G.N.; Soheilian, F.; Nagashima, K.; Veatch, S.L.; Ono, A. Roles played
by capsid-dependent induction of membrane curvature and Gag-ESCRT interactions in tetherin
recruitment to HIV-1 assembly sites. J. Virol. 2013, 87, 4650–4664.
71. Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.;
Schneider, D.K.; Nagashima, K.; et al. 3D visualization of HIV transfer at the virological synapse
between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341.
72. Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.;
Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host
Microbe 2013, 14, 468–480.
73. Wurm, C.A.; Neumann, D.; Lauterbach, M.A.; Harke, B.; Egner, A.; Hell, S.W.; Jakobs, S.
Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions
and exhibits an inner-cellular gradient. Proc. Natl. Acad. Sci. USA 2011, 108, 13546–13551.
74. Williamson, C.D.; DeBiasi, R.L.; Colberg-Poley, A.M. Viral product trafficking to mitochondria,
mechanisms and roles in pathogenesis. Infect. Disord. Drug Targets 2012, 12, 18–37.
2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Source: http://www.ecb.epm.br/~ramortara/bcel/Renato/3-%20Superresolution%20Imaging%20of%20Human%20Cytomegalovirus%20vMIA%20Localization%20in%20Sub-Mitochondrial%20Compartments-viruses-06-01612.pdf
Frozen adipose-derived mesenchymal stem cells maintain high capability to grow and differentiate
Contents lists available at adipose-derived mesenchymal stem cells maintain highcapability to grow and differentiate q Minonzio , Mattia Corazza Luca Mariotta , Mauro Gola , Michele Zanzi , Eugenio Gandolfi Domenico De Fazio Gianni Soldati Swiss Stem Cell Foundation, In Pasquée, 6925 Gentilino, SwitzerlandMolecular Diagnostic Laboratory, In Pasquée, 6925 Gentilino, SwitzerlandCentre de Chirurgie Plastique, Lausanne, SwitzerlandPlastic Surgery, Academia Day Clinic, Chiasso, SwitzerlandVia Visconti di Modrone 8/10, Milano, Italy
International journal of pharmaceutical compounding
Clinical Nuggets and Pearls: Chronic Neuropathic Painand Opioid Tolerance Marty Jones, BS Pharm, FACA, FIACP gardless of the degree, resolves when tissues tem (CNS), but chronic pain is primarily Professional Compounding Centers of heal and is usually a short-term problem. the result of a neuropathic injury or mod-