Chapter 14 - methods to determine mrna half-life in saccharomyces cerevisiae

Provided for non-commercial research and educational use only.
Not for reproduction, distribution or commercial use.
This chapter was originally published in the book Methods in Enzymology, Vol. 448, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution's administrator.
All other uses, reproduction and distribution, including without limitation commercial
reprints, selling or licensing copies or access, or posting on open internet sites, your
personal or institution's website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at:
From: Jeff Coller, Methods to Determine mRNA Half-Life in
Saccharomyces cerevisiae. In Lynne E. Maquat and Megerditch Kiledjian, editors:
Methods in Enzymology, Vol. 448, Burlington: Academic Press, 2008, pp. 267-284.
ISBN: 978-0-12-374378-7
Copyright 2008 Elsevier Inc.
Author's personal
Author's
personal
Methods to DeterminemRNA Half-Life inSaccharomyces cerevisiae
Contents1. Introduction
2. The Use of Inducible Promoters
2.1. The GAL1 UAS
2.2. A primer on galactose metabolism
2.3. Transcriptional shut-off
2.4. Transcriptional pulse-chase
2.5. Differences between transcriptional shut-off and
2.6. A GAL promoter bonus
2.7. The TET-off system
3. Measuring mRNA Decay by Use of Thermally Labile Alleles of
RNA Polymerase II
4. Measuring mRNA Decay with Thiolutin
5. RNA Extractions
5.1. Recipe for LET
5.2. RNase H cleavages of 30 UTRs
5.3. 10! Hybridization mix
5.4. 2! RNase H buffer
6. Northern Blot Analysis
7. Loading Controls
8. Determination of mRNA Half-Lives
9. Concluding Remarks
Center for RNA Molecular Biology, Case Western Reserve University, School of Medicine, ClevelandOhio, USA
Methods in Enzymology, Volume 448
# 2008 Elsevier Inc.
ISSN 0076-6879, DOI: 10.1016/S0076-6879(08)02614-1
All rights reserved.
Author's personal copy
AbstractMuch of our understanding of eukaryotic mRNA decay has come from studies inbudding yeast, Saccharomyces cerevisiae. The facile nature of genetic andbiochemical manipulations in yeast has allowed detailed investigations intothe mRNA decay pathway and the identification of critical factors required foreach step. Discussed herein are standard protocols for measuring mRNA half-lives in yeast. It should be noted, however, that variations of these assays arepossible. In addition, a few a priori considerations are addressed.
The degradation of mRNA is a vital aspect of gene regulation.
Shutting off mRNA expression ensures that previously transcribedmRNAs do not translate ad infinitum. Moreover, mRNA degradation isused by the cell as a site of regulatory responses (reviewed in ). Last, specialized mRNA turnover systems exist that recog-nize and degrade aberrant mRNAs, thereby increasing the quality control ofmRNA biogenesis (reviewed in ). Given thesefunctions, it is important to understand how decay rates of differentmRNAs are controlled and how aberrant mRNAs are targeted for destruc-tion. Much can be learned, therefore, by determining the mechanism ofhow a specific transcript is degraded under various biologic conditions or indistinct genetic backgrounds.
Polyadenylated mRNAs can be degraded in eukaryotic cells by two
general pathways (). In both cases, the degradation of the transcriptbegins with the shortening of the poly(A) tail at the 30-end of the mRNA(reviewed in ). In yeast, shortening of the poly(A)tail primarily leads to removal of the 50-cap structure (decapping), therebyexposing the transcript to digestion by a 50 to 30-exonuclease ().
mRNAs can also be degraded in a 30 to 50-direction after deadenylation
((30 to 50-degradation of mRNAs iscatalyzed by the exosome ), which is a large complex of 30 to 50-exonucleasesfunctioning in several RNA degradative and processing events (reviewed in). For the yeast mRNAs that have been studied,the process of 30 to 50-decay is slower than decapping and 50 to 30-decayHowever, it is likely that for some yeast mRNAs,or in other eukaryotic cells, 30 to 50-degradation will be the primarymechanism of mRNA degradation after shortening of the poly(A) tail(e.g.,
Author's personal copy
mRNA Half-Life in S. cerevisiae
3!>5! Exo digestion
5! > 3! Exo digestion
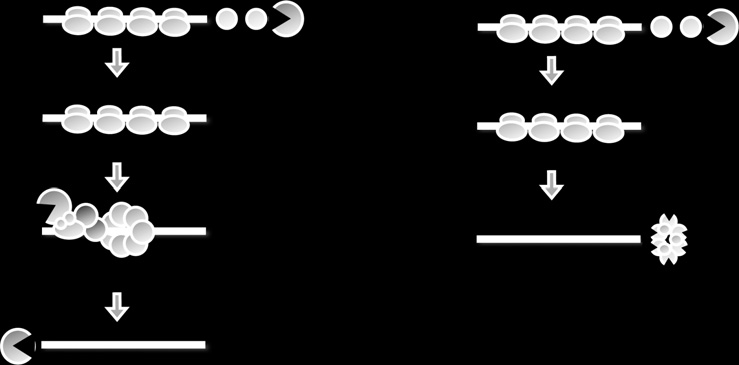
Figure 14.1 Pathways of mRNA decay in yeast. (A) Shows the major mRNA pathway
that is initiated by deadenylation, followed by removal of the 50-cap (decapping), and
lastly exonucleolytic digestion of the mRNA body in a 50 > 30-direction. (B) Degarda-
tion of mRNA can also occur by a minor pathway which is initated by deadenylation,
and then destruction of the mRNA body in a 30 >50-direction by the exosome.
In this review, I do not focus on the mechanisms of mRNA decay, but
rather, how half-life measurements are made within the cell. The techniquesI describe have proven useful in determining the steps of message decay. Itshould be noted, however, that it has been necessary to couple these assayswith genetic manipulations and other techniques to powerfully demonstratehow a particular transcript is degraded (For the assaysdescribed here, the overarching theme is the same: RNA transcription isceased in some manner, and the decay rates of specific transcripts aremonitored. The method for stopping transcription depends on the questionsasked and the information desired. I refer you to chapter 20 by Passos andParker in this issue for details on how to determine the polarity of decay andquestions related to the enzymatic activities of the decay machinery.
2. The Use of Inducible Promoters
The use of reporter mRNAs that are expressed under the control of
inducible promoters is the most common technique for measuring mRNAdecay in yeast. The preferred choice is the galactose promoter; however,others systems have been recently exploited, especially the TEToff repressiblepromoter.
2.1. The GAL1 UAS
The GAL1 upstream activating sequence (UAS) is also known as the GALpromoter or galactose inducible promoter. The GAL promoter mediatesthe expression of galactose metabolism genes, and it can confer galactose
Author's personal copy
inducibility to heterologous genes (reviewed in ). Simplyput, the GAL promoter induces mRNA expression in the presence of thesugar galactose and rapidly shuts off transcription in the presence of glucose.
This provides a powerful method for transcriptional pulse-chase and shut-off experiments in which mRNA decay kinetics is analyzed.
2.2. A primer on galactose metabolism
Before designing an RNA decay analysis with the GAL promoter, it isimportant to consider how yeasts use galactose. The GAL promoter servesas a binding site for the transcriptional regulator, Gal4p (reviewed in Gal4p has bipartite activities: a DNA binding domain, and atranscriptional activation domain. Gal4p binds strongly to the GAL UAS inthe presence of galactose, inducing transcription through its activationdomain. The addition of glucose causes a second gene, Gal80p, to bind toGal4p and mask its activation domain, thereby immediately inhibitingfurther RNA synthesis.
The choice of yeast strain is important when the GAL1 UAS is used to
control gene expression. For example, the Gal4p protein is exploited for theyeast two-hybrid system (reviewed in For this reason,yeast strains used in two-hybrid assays lack both Gal4p and Gal80p ( gal4Dand gal80D). It is not possible, therefore, to measure mRNA decay with theGAL promoter in a two-hybrid strain, because it lacks galactose-dependentregulation. It is advisable to test laboratory strains for galactose-dependentregulation before the GAL promoter is used to measure mRNA decay. Thiscan be accomplished by Northern blot analysis for the GAL1, GAL7, orGAL10 genes (see following). The yeast strains that we commonly use areBY4741 and BY4742; the WT backgrounds used in the SaccharomycesGenome Deletion Project Other strains are alsocommonly used, including S288C and W303 ).
In theory, mRNA decay of any gene can be measured with the GAL
promoter. This requires that the gene of interest be engineered to betranscribed under GAL control, either by integrating the GAL promoterinto the chromosomal gene or placing the gene on aplasmid that contains the GAL promoter ). Onceestablished, the experiment can be performed in two distinct ways: either bytranscriptional shut-off or by a transcriptional pulse-chase.
2.3. Transcriptional shut-off
Transcriptional shut-off by use of the GAL promoter is perhaps the simplestand most straightforward approach to determine decay kinetics of an mRNA.
Cells are grown in the presence of galactose until reaching mid-log phase.
Author's personal copy
mRNA Half-Life in S. cerevisiae
Because galactose is always present, the mRNA reporter is constitutivelyexpressed and at steady state when analysis is performed, which has certainramifications on the information that is gathered . Shut-off isachieved by concentrating the cells and resuspending in medium that containsglucose. Time points are then quickly taken, RNA extracted, and analysis byNorthern blot ensues. The following is a detailed protocol for this analysis:
" An overnight culture is grown in 20 ml of SGS medium ().
" The next day, 200 ml of SGS medium is inoculated overnight to an
OD600 ¼ 0.05 U/1 ml (this number is variable depending on spectro-photometer but reflects approximately 1!106 cells/mL). Grow the cellsin a 1-L flask to ensure appropriate aeration. The temperature for growthdepends on the experiment. WT strains grow best at 30 $C; mutant strainsmay only grow at 24 $C.
" Cells are harvested when reaching an OD600 ¼ 0.400 U/1 ml (3!107
cells/mL), by centrifugation in appropriately sized conical tubes.
" Working quickly, the galactose medium is poured off, and the cell pellet
is resuspended in 20 ml of S medium ). Place the culture ina small 50-ml flask in a shaking waterbath.
" 1 ml of 40% glucose (2% final concentration) is added, and time points
immediately taken.
" Use the following time course: 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, and
" Harvest each time point quickly. We use a 5 ml pipet man to remove 2 ml
of culture. Aliquots are placed in 2-ml tubes. The aliquot is quickly spundown in a small desktop picofuge, and the medium is removed byaspiration. The cell pellet is then quickly frozen on dry ice or liquidnitrogen. Ensure that the culture continues to shake in the waterbathbetween each time point.
" Once time course is complete, cell pellets can be stored at %80 $C
" Extract mRNA and analyze reporter by Northern blot analysis.
2.3.1. SGS Medium (1 L)
20 g D-galactose10 g Sucrose1.7 g Yeast nitrogen base without amino acids or ammonium acetate5.0 g Ammonium acetate2.0 g Amino acid dropout mix (variable dependent on selective
pH to 6.5 with 10 N NaOH
See for recipe.
Author's personal copy
2.3.2. S-Medium (1 L)
1.7 g Nitrogen base without amino acids or ammonium acetate5.0 g Ammonium acetate2.0 g Amino acid drop-out mix (variable dependent on selective
pH to 6.5 with 10N NaOH
2.4. Transcriptional pulse-chase
Measuring mRNA decay with transcriptional pulse-chase is a bit morechallenging, but the information obtained is exquisite. The use of a tran-scriptional pulse-chase allows for the analysis of the decay of a synchronizedpopulation of mRNA. This gives a wealth of information, because product-precursor relationships are visible (). A synchronous pool oftranscripts is made by keeping reporter expression off until just beforeanalysis. The GAL promoter is repressed by growing cells in mediumcontaining the nonfermentable carbon source raffinose. Once the appropri-ate cell density is reached, the mRNA is induced by adding galactose for abrief time, and then transcription is shut off by adding glucose. Thisprocedure generates a burst of newly synthesized transcripts whose decaycan be followed.
The following is a detailed protocol for transcriptional shut-offs:
" An overnight culture is grown in 20 ml of SR medium ().
" The next day, 200 ml of SR medium is inoculated overnight to an OD600 ¼
0.05 U/1 ml (1!106 cells/mL). Grow the cells in a1-L flask to ensureappropriate aeration. The temperature for growth depends on theexperiment.
" Cells are harvested when reaching an OD600 ¼ 0.300 U/1 ml (2.5!107
cells/mL) by centrifugation in appropriately sized conicals. For this anal-ysis, it is vital that the cell not be allowed to grow above an OD600 ¼0.300 U/1 ml. Growth above this optical density can cause spontaneousinduction of the GAL promoter ().
" Pour off medium and resuspend cell pellet in 20 ml of S medium
Place the culture in a small 50-ml flask in a shaking waterbath.
" Harvest a 2-ml aliquot to use as a preinduction control. Treat this sample
like a normal timepoint (i.e., remove medium by centrifugation and placetube on dry ice or in liquid nitrogen).
" Add 2 ml of 20% galactose (2% final concentration) and incubate culture
for 8 min in shaking waterbath.
" Separate medium from cells by centrifugation (working quickly, a 1-min.
spin at 4000 rpm is sufficient).

Author's personal copy
mRNA Half-Life in S. cerevisiae
Decapping defective strain
Transcriptional shut-off
Decapping defective strain
Figure 14.2 Comparison of a transcriptional shut-off (A). vs. a transcriptional pulse-
chase (B). The specific reporter used is the MFA2 gene under control of the GAL1UAS.
dTrepresents a sample of the RNA treated with oligo dTand RNaseH in order to indi-
cate the size of fully deadenylated RNA. PI indicates a pre-induction control. The left
panels represent analysis inWTcells, while the right panel represents an experiment per-
formed in a decapping defective strain (i.e. dcp2 mutant).
" Resuspend pellet in 20 ml of S medium, place in a 50-ml flask, and place
in shaking waterbath.
" Add 2 ml of 40% glucose (2% final concentration) and take time points
" Use the following time course: 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, and
" Harvest each time point quickly. We use a 5-ml pipet man to remove
2 ml of culture. Aliquots are placed in 2-ml tubes. The aliquot is quickly
Author's personal copy
spun down in a small desktop picofuge, and the medium is removed byaspiration. The cell pellet is then quickly frozen on dry ice or liquidnitrogen. Ensure that the culture continues to shake in the waterbathbetween each time point.
" Once time course is complete, cell pellets can be stored at %80 $C almost
" Extract mRNA and analyze reporter mRNA by Northern blot analysis
(see following).
2.4.1. SR Medium (1 L)
20 g Raffinose10 g Sucrose1.7 g Yeast nitrogen base without amino acids or ammonium acetate5.0 g Ammonium acetate2.0 g Amino acid dropout mix (variable dependent on selective requirements)1pH to 6.5 with 10 N NaOH
The biggest problem in obtaining a good transcriptional pulse-chase is
preventing preinduction of the reporter. Raffinose is a trisaccharide com-posed of galactose, fructose, and glucose. Raffinose is spontaneously hydro-lyzed at low pH to generate galactose and sucrose. It is important, therefore,to ensure the pH of the culture does not become acidic, because this wouldgenerate galactose and induce the GAL promoter. We minimize the likelihoodof spontaneous raffinose hydrolysis by adjusting our medium to pH ¼ 6.5before use. Notably, yeast synthetic medium usually has a pH of 5.0 or lower.
Second, we never allow the cell density to get above an OD600 of greaterthan 0.3 U/ml (2.5!107 cells/mL). Yeast cells secrete the enzymes invertase and
a-galactosidase. Invertase will hydrolyze raffinose into melibiose and fructose.
Melibiose is hydrolyzed by a-galactosidase into galactose and glucose, and thisinduces Gal4p-mediated gene expression ( ). Thehigher the cell density, the higher the secreted invertase and a-galactosidase levelswithin the culture, and, therefore, the higher the probability of spontaneousinduction of the GAL promoter. This has several implications for a pulse-chaseexperiment. First, cell density must be kept low enough to avoid significantconversion of raffinose but high enough to be able to obtain significantamounts of RNA from samples. Second, extracellular invertase and a-galactosi-dase accumulates in medium. Therefore, if cultures overgrow when conduct-ing a pulse-chase, they cannot be simply diluted back to a lower OD. If dilutingcultures back, it is advisable to pellet cells by centrifugation, wash with freshmedium, and then resuspend in new medium to an appropriate OD. Last, thechoice of yeast strain can also have an impact on invertase levels in the medium.
The S. cerevisiae genome contains six unlinked loci that encode invertase,SUC1-5, and SUC7 Yeast strains can carry anynumber or combination of invertase genes. For example, S288C strains only
Author's personal copy
mRNA Half-Life in S. cerevisiae
contain the SUC2 gene It is theoretically possibleto avoid preinduction by use of sucrose as the carbon source rather thanraffinose (Ambro Van Hoof 2008), although this has not been tested rigorously.
2.5. Differences between transcriptional shut-off and
Both a transcriptional shut-off and a transcriptional pulse-chase will giveimportant information about the half-life of an mRNA. The differencebetween these two analyses is that a shut-off is a decay from steady state,whereas a pulse-chase monitors the decay of a synchronized population(). There are times when one analysis is preferred over the other.
Shut-off experiments are much easier than pulse-chase experiments,because cells grow better in galactose than in raffinose, and preinductionis not a concern. A transcriptional shut-off, however, gives only an approx-imation of how mRNA decay is impaired. In a transcriptional pulse-chase,the progression of a transcript through the steps of mRNA decay, (i.e.,deadenylation, decapping, and exonucleolytic digestion) can be observed.
This is especially important when trying to determine what step in decay isaffected by an experimental condition ().
2.6. A GAL promoter bonus
One particular advantage of choosing to use the GAL promoter for mRNAdecay analysis is that, besides the reporter GAL UAS fusion, it is also possibleto monitor the decay of three endogenous mRNAs: the GAL1, GAL7, andGAL10 transcripts. All three mRNAs are naturally controlled by galactoseand thus will be regulated transcriptionally like the reporter. These becomepowerful controls especially when determining transcript specific effects.
2.7. The TET-off system
Although not as common, mRNA decay has been successfully monitoredby shutting off transcription with a tetracycline-dependent repressible pro-moter. Developed by , the gene of interest is fused to thetetracycline operator (tetO). The plasmid must also express a second genecalled the tTA transactivator. The tTA transactivator consists of the VP16activator domain of herpes simplex virus fused to the tetracycline-induciblerepressor (tetR) from the Tn10-encoded tetracycline-resistance operon). Under these conditions, genes under tetO control areexpressed when tetracycline is absent, but transcription is quickly shut-offwhen the antibiotic is added to media. Commonly, doxycycline, which is aderivative of tetracycline, is used. This system is useful when trying to
Author's personal copy
control expression of a single mRNA without altering carbons sources). However, this approach is limited, becauseonly decay from the steady state can be performed.
3. Measuring mRNA Decay by Use of Thermally
Labile Alleles of RNA Polymerase II
Another useful method for analyzing mRNA decay takes advantage of
the temperature-sensitive allele rpb1-1 (The rpb1-1 allelemaps to the RPO21 gene, which encodes the largest subunit of RNApolymerase II. At the permissive temperature, rpb1-1 functions normally.
Shifting to the restrictive temperature leads to a quick and synchronousdisruption of all pol II transcription. The advantage of this approach is thatthe analysis of endogenous mRNAs can be measured. No prior cloning oralteration of promoter elements is required. In addition, decay analysis withrpb1-1 is not dependent on nutrient conditions, allowing the effects of stressor starvation to be monitored ). Last, because rpb1-1 stopsall pol II transcription, decay of any mRNAs can be monitored.
The disadvantage of rpb1-1 is that the allele must be incorporated intodifferent strains if distinct genetic backgrounds are desired. In addition,rbp1-1 shut-off experiments analyze decay from the steady state. Therefore,product-precursor relationships cannot be determined as in transcriptionalpulse-chase experiments. Nonetheless, it has proven advantageous to userpb1-1 to measure mRNA decay in some experimental settings. Thefollowing is a detailed protocol.
" An overnight culture is grown at 24 $C in 20 ml of medium (medium
choice depends on need).
" The next day, 200 ml of medium is inoculated with the overnight, and
cells are grown to an OD600 ¼ 0.05 U/1 ml (1!106 cells/mL). In a 1-Lflask to ensure appropriate aeration. Cultures must be grown at 24 $C.
" Ten milliliters of medium is preheated to 56 $C at least 1 h before
performing the shut-off and, a 50-ml flask is preheated to 37 $C in ashaking waterbath.
" Cells are harvested when reaching an OD600 ¼ 0.400 U/1 ml (3!107
cells/mL), by centrifugation in appropriately sized conicals. Resuspendcell pellet in 10 ml of room temperature medium.
" Working quickly, the preheated medium is poured into the freshly
harvested 10-ml culture. The combination of 10 ml of medium at56 $C and 10 ml of culture at 24 $C gives a rapid shift to 37 $C.
" Place the culture in the preheated 50-ml flask in a shaking waterbath set
" Immediately take time points at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50,
Author's personal copy
mRNA Half-Life in S. cerevisiae
" Harvest each time point quickly as previously described.
" Cell pellets can be stored at %80 $C indefinitely.
Extract mRNA and analyze reporter by Northern blot analysis.
4. Measuring mRNA Decay with Thiolutin
Chemical means can also be used to measure mRNA decay in yeast
). Thiolutin is a sulfur-containing antibiotic fromStreptomyces laterosporus that inhibits RNA synthesis directed by all threeyeast RNA polymerase. Adding thiolutin at low concentrations is sufficientto completely block RNA synthesis, thereby allowing mRNA decay anal-ysis. Like the use of the rpb1-1 allele, this approach allows the analysis ofendogenous mRNAs. Moreover, the effects of stress or nutrient starvationon mRNA decay can be monitored, because carbon source choice isirrelevant. Last, the decay of any RNA can be monitored because thiolutinblocks the activity of all three RNA polymerases. Unlike an rpb1-1 mutant,however, a thiolutin shut-off can be conducted on any preexisting strain;genetic engineering is not required. The disadvantage of thiolutin, like rpb1-1,is that shut-off experiments limit the analysis to decay from the steady state.
Last, new evidence suggests that some mRNAs may be stabilized by theresponse of cells to the drug ). Nonetheless,for many mRNAs, thiolutin has proven useful in monitoring decay kinetics.
The protocol is analogous to the protocols we have previously described fortranscriptional shut-off experiments except that 3 mg/ml (final concentration)of thiolutin is added before taking time points.
5. RNA Extractions
Irrespective of how mRNA decay analysis is performed, once samples
are collected, it is necessary to extract the mRNA and analyze decay rates.
The following is our protocol for extracting total-cell mRNA from yeast.
We analyze total RNA by Northern blotting. Because many mutations andconditions can affect the decapping step exclusively, it is important to notpoly(A)-select mRNA by use of oligo(dT) columns before analysis. Altera-tions in decapping stabilize poly(A)-minus mRNAs. Therefore oligo(dT)selection would result in the loss of information.
" Resuspend frozen yeast cell pellets (in 2-ml tube) in 150 ml LET
" Add 150 ml phenol equilibrated with LET.
Author's personal copy
" Add equal volume (approximately 300 to 400 ml) of glass beads (bead size ¼
500 mm; available from Sigma-Aldrich; cat #G8772).
" Vortex in MultiMixer (VWR cat# 58816-115) for 5 min at top speed.
" Add 250 ml DEPC H2O and 250 ml 1:1 phenol/chloroform equilibrated
" Vortex in MultiMixer for an additional 5 min followed by centrifugation
for 5 min at 14,000 rpm.
" Transfer aqueous phase (approximately 450 ml, top layer) to new 1.5-ml
" Add 450 ml of 1:1 phenol/chloroform equilibrated with LET. Vortex 60 sec
and spin 3 min at 14,000 rpm.
" Transfer aqueous phase to new 1.5-ml tube.
" Add 400 ml of chloroform. Vortex 60 sec and spin 3 min at 14,000 rpm.
" Transfer aqueous phase to a new 1.5-ml tube. Add 40 ml of 3 M sodium
acetate and 800 ml of 100% cold ethanol. Mix well and place at %20 $Cfor 1 h.
" Collect RNA by centrifugation at room temperature for 10 min at
14,000 rpm. Wash pellet with 500 ml 70% EtOH and recentrifuge for 5min. Drain supernatant and dry pellet (either air-dry or dry in SpeedVac[no heat]).
" Resuspend pellet in 50 to 150 ml DEPC dH2O.
" Quantify each RNA sample by measuring the absorbance at 260 nm with
spectrophotometer. Assay 4 ml of RNA in 996 ml of dH2O.
" Determine concentration of each sample on the basis of the extinction
coefficient for RNA of 40 mg/ml. (Note: if the preceding dilution is used,simply multiply the OD260 by 10; this equals the concentration of eachsample in mg/ml).
" Analyze 10 to 30 mg of each sample by Northern blot.
5.1. Recipe for LET
25 mM Tris, pH 8.0100 mM LiCl20 mM EDTA(All reagents should be made in DEPC-treated distilled H2O).
5.2. RNase H cleavages of 30 UTRs
mRNA decay in yeast is initiated by deadenylation (reviewed in The rate of poly(A) shortening, therefore, is an importantconsideration in decay analysis. The poly(A) tail typically ranges in size from10 to 100 adenosines. Because the tail is small, the size difference betweenadenylated and deadenylated transcripts is negligible for most large mRNAs.
It is necessary, therefore, to first cleave an mRNA within its 30 UTR with
Author's personal copy
mRNA Half-Life in S. cerevisiae
an antisense oligonucleotide and RNase H to generate a smaller RNAfragment that can be analyzed by high-resolution Northern blotting afterelectrophoresis in polyacrylamide gels. The antisense oligo that we typicallyuse is 20 nucleotides in size and binds near the stop codon of the gene ofinterest. The following is the protocol for generating a smaller mRNAfragment from full-length mRNA.
" Dry down in SpeedVac 10 mg of total-cell RNA and 300 ng antisense
oligo (can dry down as much as 40 mg of RNA and still maintain the300 ng of oligo; do not overdry, because it will be difficult to resuspend).
To control for poly(A) tail lengths, also treat a sample with antisense oligoand 300 ng of oligo d(T).
" Resuspend pellet in 10 ml of 1! hybridization mix (
" Heat sample at 68 $C for 10 min. Cool slowly to 30 $C. Pulse spin down.
" Add 9.5 ml of 2! RNase H buffer and 0.5 ml RNase H. Mix well.
" Incubate sample at 30 $C for 60 min.
" Add 180 ml stop mix. Extract sample with 200 ml of phenol/chloroform.
Remove aqueous phase and extract with 200 ml of chloroform.
" Precipitate RNA by adding 500 ml of 100% ethanol. Freeze at %20 $C for
" Spin down sample for 10 min at room temperature. Wash RNA pellet
with 300 ml of 70% ethanol. Dry either at room temperature or undervacuum in Speedvac (once again, do not overdry).
" Resuspend sample in 10 ml of DEPC-treated water and add 10 ml of
loading dye. Heat sample at 100 $C for 5 min before loading in 6 to 8%denaturing acrylamide gel. After resolving by PAGE, perform a NorthernBlot analysis and detect RNA fragment with an mRNA-specific probe.
" Prepare solutions with DEPC-treated distilled water and store in aliquots
5.3. 10! Hybridization mix0.25 M Tris-HCl, pH 7.510 mM EDTA0.5 M NaCl
5.4. 2! RNase H buffer40 mM Tris-HCl, pH 7.520 mM MgCl2100 mM NaCl2 mM DTT60 mg/ml BSA
Author's personal copy
0.04 mg/ml tRNA20 mM EDTA300 mM NaOAc
6. Northern Blot Analysis
After performing an mRNA decay analysis, it is possible to obtain a
half-life measurement by resolving mRNA in a formaldehyde agarose gelfollowed by Northern blot. To obtain information about the deadenylationrate, it is necessary to analyze mRNA or RNase H fragments by resolutionin a 6% polyacrylamide gel followed by Northern blotting. Protocols forthese techniques are described elsewhere and Chap-ter 17 by Chen et al. in this volume).
7. Loading Controls
It is important to consider several controls in an mRNA decay analysis.
First, if monitoring deadenylation rate, a sample of mRNA (usually from thefirst time point) should be treated with RNase H and oligo (dT) to generate anonadenylated marker that indicates the size of the fully deadenylatedmRNA (). Second, the Northern blot should be reprobedwith a loading control. SCR1 RNA is ideal for this purpose. SCR1 RNA isthe 7S signal recognition particle RNA used by the cell for cotranslationalprotein targeting to the endoplasmic reticulum. Importantly, this small522-nt RNA is noncoding and transcribed by RNA polymerase III and isunaffected by changes in mRNA transcription except in the case of thiolutin.
Thiolutin inhibits all three RNA polymerases, including SCR1 RNA levels,albeit only slightly within the recommended 1 h time course. SCR1 RNA isabundant and can be easily detected with a gamma 32P-ATP kinase-labeledoligonucleotide probe.
8. Determination of mRNA Half-Lives
Yeast mRNAs generally decay with first-order kinetics
Half-lives, therefore, can be represented by the equationt½ ¼ 0.693/k, where k ¼ the rate constant for mRNA decay (i.e., percentchange over time). Half-life calculations for a particular mRNA transcript

Author's personal copy
mRNA Half-Life in S. cerevisiae
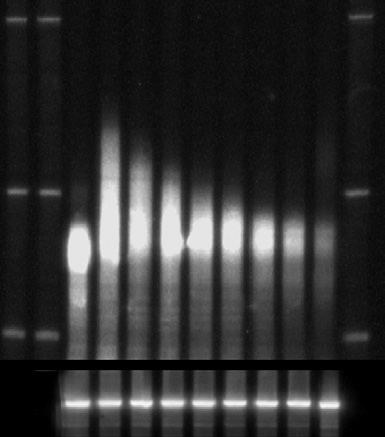
Transcriptional shut-off analyzes of the PGK1mRNA (A) and calculation
of PGK1half-life (B). In (A) the mRNA reporter was cleaved by RNaseH using an anti-
sense oligo and then resolved by PAGE. dT represents a sample that was also cleaved
with oligo d(T) and RNaseH to indicate size of fully deadenylated mRNA. Below is a
Northern of the SCR1 RNAwhich is used as a loading control. (B) is the quantitation
of the experiment shown in (A) and representation of these data on a semi-log plot.
This was used to calculate the half-life based on the protocol outlined in the text.
Author's personal copy
are made by quantitating the amount of mRNA present at each time pointwith a PhosphorImager. This value should be normalized for loadingvariations with a control like SCR1 RNA (). The value for kcan be determined with a semilogarithmic plot of the concentration ofmRNA over time and determining the slope of a best-fit line (slope¼k;Once the rate constant is determined, the half-life can becalculated. It is strongly advised to measure mRNA half-lives at least threetimes to ensure reproducibility.
9. Concluding Remarks
In summary, the techniques described in this review provide useful
methods for measuring mRNA decay rates in yeast. The most importantconsideration in choosing a particular technique is to determine the type ofinformation that is desired. If simple half-life measurements are needed,then a transcriptional shut-off experiment will suffice. If information issought about the sub-step of decay affected, then a transcriptional pulse-chase is required.
The techniques described are amalgamated from the work of many laboratories and indivi-duals, especially Drs. Roy Parker, Allan Jacobson, Stuart Peltz, Denise Muhlrad, CarolineDecker, David Herrick, Kristian Baker, Ambro Van Hoof, Pat Hilleren, and RichardYoung. I also thank Drs. Wenqian Hu, Thomas Sweet, and Sarah Geisler for advice andcriticism of the manuscript. Funding is provided by National Institutes of Health(GM080465).
Anderson, J. S., and Parker, R. P. (1998). The 30 to 50-degradation of yeast mRNAs is a
general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and30 to 50-exonucleases of the exosome complex. EMBO J. 17, 1497–1506.
Baker, K. E., and Parker, R. (2004). Nonsense-mediated mRNA decay: Terminating
erroneous gene expression. Curr. Opin. Cell Biol. 16, 293–299.
Burke, D., Dawson, D., and Stearns, T. (2000). In ‘‘Methods in yeast genetics: A Cold
Spring Harbor Laboratory Course Manual,'' Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
Cao, D., and Parker, R. (2001). Computational modeling of eukaryotic mRNA turnover.
RNA 7, 1192–1212.
Carlson, M., and Botstein, D. (1982). Two differentially regulated mRNAs with different
50-ends encode secreted with intracellular forms of yeast invertase. Cell 28, 145–154.
Coller, J., and Parker, R. (2004). Eukaryotic mRNA decapping. Annu. Rev. Biochem. 73,
Author's personal copy
mRNA Half-Life in S. cerevisiae
Coller, J., and Parker, R. (2005). General translational repression by activators of mRNA
decapping. Cell 122, 875–886.
Decker, C. J., and Parker, R. (1993). A turnover pathway for both stable and unstable
mRNAs in yeast: Evidence for a requirement for deadenylation. Genes Dev. 7,1632–1643.
Gari, E., Piedrafita, L., Aldea, M., and Herrero, E. (1997). A set of vectors with a
tetracycline-regulatable promoter system for modulated gene expression in Saccharomycescerevisiae. Yeast 13, 837–848.
He, F., and Jacobson, A. (1995). Identification of a novel component of the nonsense-
mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev. 9,437–454.
Herrick, D., Parker, R., and Jacobson, A. (1990). Identification and comparison of stable and
unstable mRNAs in Saccharomyces cerevisiae. Mol. Cell. Biol. 10, 2269–2284.
Higgs, D. C., and Colbert, J. T. (1994). Oat phytochrome A mRNA degradation appears to
occur via two distinct pathways. Plant Cell 6, 1007–1019.
Hilgers, V., Teixeira, D., and Parker, R. (2006). Translation-independent inhibition of
mRNA deadenylation during stress in Saccharomyces cerevisiae. RNA 12, 1835–1845.
Hilleren, P. J., and Parker, R. (2003). Cytoplasmic degradation of splice-defective pre-
mRNAs and intermediates. Mol. Cell 12, 1453–1465.
Hsu, C. L., and Stevens, A. (1993). Yeast cells lacking 50!30-exoribonuclease 1 contain
mRNA species that are poly(A) deficient and partially lack the 50-cap structure. Mol. CellBiol. 13, 4826–4835.
Johnston, S. A., and Hopper, J. E. (1982). Isolation of the yeast regulatory gene GAL4 and
analysis of its dosage effects on the galactose/melibiose regulon. Proc. Natl. Acad. Sci. USA79, 6971–6975.
Lohr, D., Venkov, P., and Zlatanova, J. (1995). Transcriptional regulation in the yeast GAL
gene family: A complex genetic network. FASEB J. 9, 777–787.
Longtine, M. S., McKenzie, A., 3rd, Demarini, D. J., Shah, N. G., Wach, A., Brachat, A.,
Philippsen, P., and Pringle, J. R. (1998). Additional modules for versatile and economicalPCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14,953–961.
Muhlrad, D., Decker, C. J., and Parker, R. (1994). Deadenylation of the unstable mRNA
encoded by the yeast MFA2 gene leads to decapping followed by 50!30 digestion of thetranscript. Genes Dev. 8, 855–866.
Muhlrad, D., Decker, C. J., and Parker, R. (1995). Turnover mechanisms of the stable yeast
PGK1 mRNA. Mol. Cell Biol. 15, 2145–2156.
Mukherjee, D., Gao, M., O'Connor, J. P., Raijmakers, R., Pruijn, G., Lutz, C. S., and
Wilusz, J. (2002). The mammalian exosome mediates the efficient degradation ofmRNAs that contain AU-rich elements. EMBO J. 21, 165–174.
Nonet, M., Scafe, C., Sexton, J., and Young, R. (1987). Eucaryotic RNA polymerase
conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell Biol. 7, 1602–1611.
Pelechano, V., and Perez-Ortin, J. E. (2007). The transcriptional inhibitor thiolutin blocks
mRNA degradation in yeast. Yeast.
Rodgers, N. D., Wang, Z., and Kiledjian, M. (2002). Characterization and purification of a
mammalian endoribonuclease specific for the alpha-globin mRNA. J. Biol. Chem. 277,2597–2604.
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). ‘‘Molecular Cloning: A Laboratory
Manual,'' 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Traven, A., Jelicic, B., and Sopta, M. (2006). Yeast Gal4: A transcriptional paradigm
revisited. EMBO Rep. 7, 496–499.
Author's personal copy
van Hoof, A., and Parker, R. (2002). Messenger RNA degradation: Beginning at the end.
Curr. Biol. 12, R285–287.
Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B.,
Bangham, R., Benito, R., Boeke, J. D., Bussey, H., et al. (1999). Functional characteri-zation of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285,901–90.
Source: http://www.case.edu/med/coller/Coller%20MIE%202009.pdf
yorkleighsurgery.co.uk
Information For Patients 93 st George's road, Cheltenham, Gloucestershire Gl50 3eD Appointments: (01242) 255337 • Tel: (01242) 519049 fax: (01242) 253556 Urgent out-of-hours service: 0300 4210 220 Surgery Opening Hours The Surgery Is Open .Monday - Friday 8.00am - 6.30pm Extended Opening Hours .Monday Evening 6.30 - 8.00pm
Chapter 14 veterinary aspects
COMMISSIONED PAPER (UK) This paper was commissioned by FECAVA for the Special issue of EJCAP, Genetic/Hereditary Disease and Breeding. Must not be copied without permission © 2014 Chiari–like malformation and syringomyelia Clare Rusbridge Introduction Syringomyelia is a condition characterised by fluid filed cavities (syrinxes or syringes) within the central spinal