International.insel.ch

The leukemic fusion gene AML1-MDS1-EVI1
suppresses CEBPA in acute myeloid leukemia by
activation of Calreticulin
Daniel Helbling*, Beatrice U. Mueller†, Nikolai A. Timchenko‡, Anne Hagemeijer§, Martine Jotterand¶,
Sandrine Meyer-Monard储, Andrew Lister**, Janet D. Rowley††, Barbara Huegli*, Martin F. Fey*, and Thomas Pabst*‡‡
*Institute of Medical Oncology and †Department of Medicine, University Hospital, CH-3010 Bern, Switzerland; ‡Department of Pathology and HuffingtonCenter on Aging, Baylor College of Medicine, Houston, TX 77030; §Center for Human Genetics, University of Leuven, B-3000 Leuven, Belgium; ¶DivisionAutonome de Ge´ne´tique Me´dicale, Centre Hospitalier Universitaire Vaudois, CH-1011 Lausanne, Switzerland ; 储Division of Hematology, University Hospital,CH-4031 Basel, Switzerland; **Department of Medical Oncology, St. Bartholomew's Hospital, London EC1A 7BE, United Kingdom; and ††Section ofHematology兾Oncology, Department of Medicine, University of Chicago Medical Center, Chicago, IL 60637
Contributed by Janet D. Rowley, July 27, 2004
The leukemic fusion gene AML1-MDS1-EVI1 (AME) encodes a
program (25, 27). In addition, no mature granulocytes are observed
chimeric transcription factor that results from the t(3,21)(q26;q22)
in cebpa knock-out mice, whereas all other blood-cell types are
translocation seen in patients with acute myeloid leukemia, with
present in normal numbers (24).
therapy-related myelodysplastic syndrome, or with chronic my-
We showed previously that dominant-negative mutations of the
eloid leukemia in blast crisis. The myeloid transcription factor
CEBPA gene are found in a significant proportion of patients with
CEBPA is crucial for normal granulopoiesis. Here, we found that
myeloblastic subtypes (M1 and M2) of AML (29–31). Further-
conditional expression of AME suppresses CEBPA protein by 90.8%
more, we demonstrated that the AML1-ETO fusion protein sup-
and DNA-binding activity by 93.9%. In contrast, CEBPA mRNA
presses CEBPA expression (32). Here, we found that AME sup-
levels remained unchanged. In addition, we detected no differ-
presses CEBPA protein; in contrast to the AML1-ETO fusion, it
ences in CEBPA mRNA levels in leukemic blasts of patients carrying
fails to suppress CEBPA mRNA expression. We identified trans-
the AME translocation (n ⴝ 8) compared to acute myeloid leukemia
lational inhibition of CEBPA mediated by induction of calreticulin
patients with a normal karyotype (n ⴝ 9). CEBPA protein and
(CRT), a ubiquitous protein with calcium storage and chaperone
binding activity, however, were reduced significantly (100% and
function, as a mechanism involved in leukemia.
92.1%, respectively) in AME patient samples. Furthermore, we
observed that calreticulin (CRT), a putative inhibitor of CEBPA
Materials and Methods
translation, was strongly activated after induction of AME in the
Patient Samples. Ficoll-separated, fresh, mononucleated peripheral
cell-line system (14.8-fold) and in AME patient samples (12.2-fold).
blood or bone marrow cells of AML patients were collected at the
Moreover, inhibition of CRT by small interfering RNA powerfully
time of diagnosis before initiation of treatment. Conventional
restored CEBPA levels. These results identify CEBPA as a key target
cytogenetic analysis was performed in each patient (Table 1).
of the leukemic fusion protein AME and suggest that modulation
of CEBPA by CRT may represent a mechanism involved in the
Generation of Cell Line with Conditional AME Expression. The U937T
differentiation block in AME leukemias.
cell line with the tetracycline transactivator under the control of a
tetracycline-responsive element was obtained from Gerard Gros-
Acute myeloid leukemia (AML) is a clonal malignant disease veld (St. Jude Children's Research Hospital, Memphis, TN). A
characterized by a block in normal myeloid differentiation
4.0-kb ScaI兾XbaI fragment of the pcDNA3 vector, containing the
leading to the accumulation of immature hematopoietic cells in the
neomycin resistance gene, was ligated with the 0.95-kb ScaI兾XbaI
bone marrow and peripheral blood (1). AML is characterized
fragment of the tetracycline-off response plasmid pTRE. A 5.2-kb
further by the presence of specific balanced chromosome rear-
EcoR1兾XbaI fragment encoding for the entire AME cDNA was
rangements that create novel fusion genes (2). However, little is
introduced into the pTRE-neo plasmid. The plasmid was trans-
known about the mechanisms of how such fusion genes contribute
fected into U937T cells by electroporation. Eighteen single-cell
to the differentiation block.
clones were tested for AME induction. The clone with the maximum
AML1-MDS1-EVI1 (AME) is a chimeric fusion gene observed in
increase of AME mRNA transcripts was selected for additional
patients with de novo or therapy-related AML, with therapy-related
myelodysplastic syndrome (MDS), or with chronic myeloid leuke-
mia in blast crisis (CML-BC) (3–5). AME is an in-frame fusion of
Real-Time PCR and Sequencing. For isolation of total RNA, the
the AML1 and MDS1兾EVI1 genes (6). AML1 (also known as
RNeasy minikit (Qiagen, Hilden, Germany) was used. Real-time
RUNX1) is one of the most frequently translocated or mutated
PCR was performed on the ABI PRISM 7700 sequence-
genes in human cancer (7–12). EVI1 is abnormally expressed in
detection system by using TaqMan Universal PCR Master Mix.
human MDS, AML, and CML-BC that are associated with the
For CEBPA and CRT mRNA quantitation by Assays-on-
t(3,3)(q21q26) or inv(3)(q21q26) (3, 13, 14). MDS1 is a gene of
Demand gene-expression probes (Applied Biosystems) were
largely unknown function located upstream of EVI1. Mice trans-
used. Primers for AME detection were targeting the AML1兾
planted with syngeneic bone marrow cells expressing the AME
MDS1 transition (Assays-by-Design gene-expression probes, Ap-
fusion gene develop a disease similar to human acute myelomono-
plied Biosystems). The primers were 5⬘-AACCACTCCACT-
cytic leukemia (15). In addition, AME has been shown to induce
proliferation and inhibit differentiation in myeloid cells (16, 17).
CEBPA plays distinct roles in the differentiation process of
Abbreviations: AML, acute myeloid leukemia; AME, AML1-MDS1-EVI1; MDS, myelodys-plastic syndrome; CML, chronic myeloid leukemia; CML-BC, CML in blast crisis; CRT, calre-
various cell types (18–25). In the hematopoietic system, CEBPA is
ticulin; G-CSF, granulocyte colony-stimulating factor; siRNA, small interfering RNA; CML-
expressed exclusively in myelomonocytic cells (18, 25). Conditional
CP, chronic phase of CML.
expression of CEBPA is sufficient to trigger terminal neutrophil
‡‡To whom correspondence should be addressed. E-mail: [email protected].
differentiation (25–28) and block the monocytic differentiation
2004 by The National Academy of Sciences of the USA
13312–13317 兩 PNAS 兩 September 7, 2004 兩 vol. 101 兩 no. 36

Table 1. Clinical presentation of patients
M, male; F, female; FAB, French-American-British classification; WBC, white blood cell count; PBL, peripheral
blood leukocytes; LDH, lactate dehydrogenase; G兾I: 109 per liter.
GCCT-3⬘ and 5⬘-ATACCGTTGATGGGACTTTATGGAAA-
3⬘, containing CCG repeats) were annealed. The double-stranded
3⬘, and the probe was 5⬘-FAM-CAGTCTACGTCTTACT-
oligomers were separated from single-stranded oligomers by gel
TAMRA-3⬘ (FAM, 6-carboxyfluorescein; TAMRA, N,N,N⬘,N⬘-
electrophoresis and subsequent extraction. The double-stranded
tetramethyl-6-carboxyrhodamine). 7S was used as reference
oligomers were labeled by using [␥-32P]ATP, and equal amounts
gene. N-fold changes were calculated as: n-fold ⫽ (Ct1 ⫺ Ct2)2
were incubated with whole-cell protein extracts for 30 min at room
⫻ PCR efficiency (Ct, cycle threshold). The PCR efficiency was
temperature and subjected to UV treatment for 5 min at 125 mJ
calculated based on a standard curve. Sequencing of the CEBPA
(34). After electrophoresis, the proteins were transferred to the
gene was done as described (29).
membrane and autoradiographed.
Western Blot Analysis. CEBPA, CEBPB, CEBPE, granulocyte col-
RNA Interference. CRT small interfering RNA (siRNA) (Ambion,
ony-stimulating factor (G-CSF) receptor, AME, and CRT proteins
Austin, TX) had the sequences 5⬘-GGAGCAGUUUCUGGACG-
were detected with rabbit polyclonal antibody against CEBPA
GATT-3⬘ and 5⬘-UCCGUCCAGAAACUGCUCCTT-3⬘. As con-
(1:500; Santa Cruz Biotechnology), a rabbit polyclonal antibody
trol, the Silencer negative control no. 2 siRNA (Ambion) was used.
against CEBPB (1:1,000; Santa Cruz Biotechnology), a rabbit
U937 AME cells were set to a density of 1.4 ⫻ 106 in 100 l of
polyclonal antibody against CEBPE (1:1,000; Santa Cruz Biotech-
Amaxa solution V (Nucleofector kit V, Amaxa, Cologne, Germany)
nology), a rabbit polyclonal antibody against G-CSF receptor
and mixed with 800 ng of siRNA. Cells were transfected by
(1:500; Santa Cruz Biotechnology), a rabbit polyclonal antibody
electroporation applying NUCLEOFECTOR TECHNOLOGY (software
against AML1B (1:500; Oncogene Science), and a rabbit polyclonal
version 2.1, Amaxa, Gaithersburg, MD).
antibody against CRT (1:200,000; Sigma) followed by an IgG-
horseradish peroxidase-conjugated secondary antibody against rab-
Statistical Analysis. Mean and SD were calculated. Statistical anal-
bit (Amersham Pharmacia Biosciences). A monoclonal anti-rabbit
ysis was performed by using the Mann–Whitney rank sum test
-actin antibody served as a loading control (Sigma).
(SIGMASTAT 3.0).
Electrophoretic Mobility Shift Assays. The G-CSF receptor promotor
oligonucleotide (bp ⫺57 to ⫺38) had the sequence 5⬘-
Conditional Expression of AME Suppresses CEBPA Protein in U937
AAGGTGTTGCAATCCCCAGC-3⬘ (the CEBP-binding site is
Leukemic Cells. We established single-cell clones of the myeloid
underlined). An electrophoretic mobility shift assay was performed
leukemic cell line (U937) that conditionally express the AME
as described (25, 29, 32, 33). Quantitative CEBPA- and CEBPB-
protein after withdrawal of tetracycline. Real-time PCR analysis
binding activity was assessed further by using an ELISA-based assay
showed in 4 of 18 clones a ⬎10-fold increase of AME mRNA
(TransAM, Active Motif, Carlsbad, CA). Briefly, a 96-well plate
transcripts 48 h after withdrawal of tetracycline. No morphological
was coated with the immobilized oligo 5⬘-CTTGCGCAATC-
changes were observed after the induction of AME (data not
TATA-3⬘ (the CEBP consensus binding site is underlined). Nuclear
shown). The clone selected for the experiments in this study showed
extracts were added together with a CEBPA antibody. Addition of
an increase in AME mRNA expression of 25-fold on day 2 (Fig.
a secondary antibody conjugated to horseradish peroxidase pro-
1A). However, no CEBPA mRNA changes were observed (n-fold
vided colorimetric quantitation by spectrophotometry.
range, 1.03–1.37; three independent experiments) (Fig. 1A). On day
2, the median absolute cycle threshold values for AME and CEBPA
UV Cross-Link Assay for CRT. A double-stranded RNA oligomer
were 21.7 (SD, 0.1) and 18.4 (SD, 0.3), respectively. We thus
covering a CRT-binding site within the CEBPA mRNA was gen-
concluded that forced expression of AME had no effect on CEBPA
erated as follows: oligomer A (5⬘-CCCCACGGGCGGCGGCG-
mRNA levels.
GCGGCGGCGACUU-3, containing CGG repeats) and oligomer
Western blot analysis verified the induction of the 200-kDa AME
protein (Fig. 1B). In contrast to CEBPA mRNA levels, CEBPA
Helbling et al.
PNAS 兩 September 7, 2004 兩 vol. 101 兩 no. 36 兩 13313

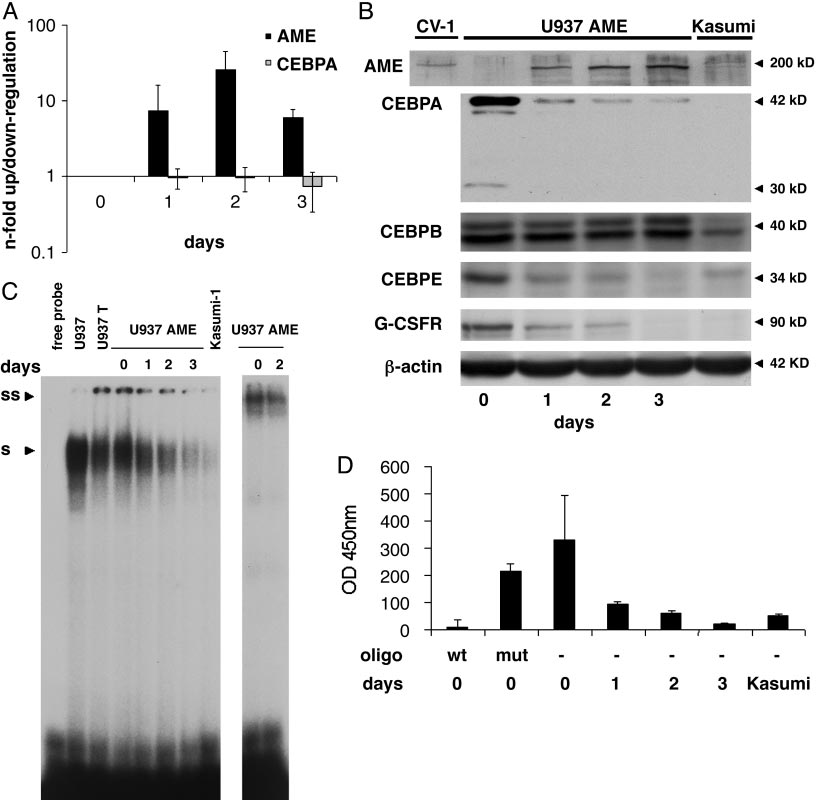
Conditional expression of AME in U937 leukemic cells. (A) U937 cells were analyzed before (day 0) and 1, 2, and 3 days after withdrawal of tetracycline
by real-time PCR analyses for AME and CEBPA expression. Mean values and SD (error bars) are depicted. (B) Western blot analyses at the same time points as inA. CV1 cells transiently transfected with an AME expression construct served as positive control (left lane). The membrane was incubated further with antibodiesagainst CEBPA, CEBPB, CEBPE, G-CSF receptor, and -actin. (C) CEBPA-binding activity to a CEBP site as present in the G-CSF receptor promoter was assessed byelectrophoretic mobility shift assays at the time points indicated after withdrawal of tetracycline. Kasumi-1 cells served as a CEBPA negative control. S, shiftedCEBPA protein; SS, supershifted CEBPA-protein complex. (D) CEBPA-binding activity measured with the TransAM assay. A CEBP wild-type (wt) or a CEBP mutatedoligonucleotide (mut) were added to nuclear extracts from day 0 as a control. Mean values and SD (error bars) are depicted. Nuclear extracts from Kasumi-1 cellsserved as a negative control.
protein was rapidly suppressed after AME induction (Fig. 1B). The
24 h after AME induction, we observed a consistent decrease of
blot was subsequently incubated with antibodies against other
CEBPA binding to this site. At day 3, binding activity was hardly
CEBP family members and the G-CSF receptor (Fig. 1B). CEBPB
detectable (Fig. 1C), which is equivalent to a 93.6% reduction of
protein remained unchanged after AME induction, consistent with
binding activity as further verified by the TransAM assay (Fig. 1D).
findings after AML1-ETO induction (32). CEBPE is reported to be
In contrast, we observed no changes in DNA-binding activity in the
a downstream target of CEBPA (29, 32). As expected, we observed
parental U937T cells after withdrawal of tetracycline (data not
a marked decrease in CEBPE protein after AME induction. In
shown). In conclusion, these experiments confirm that AME
addition, the G-CSF receptor protein as another direct target of
indeed suppresses the CEBPA-protein production and function.
CEBPA (24, 32, 33) was similarly suppressed after AME induction.
To ensure that the induction of the tetracycline system itself had no
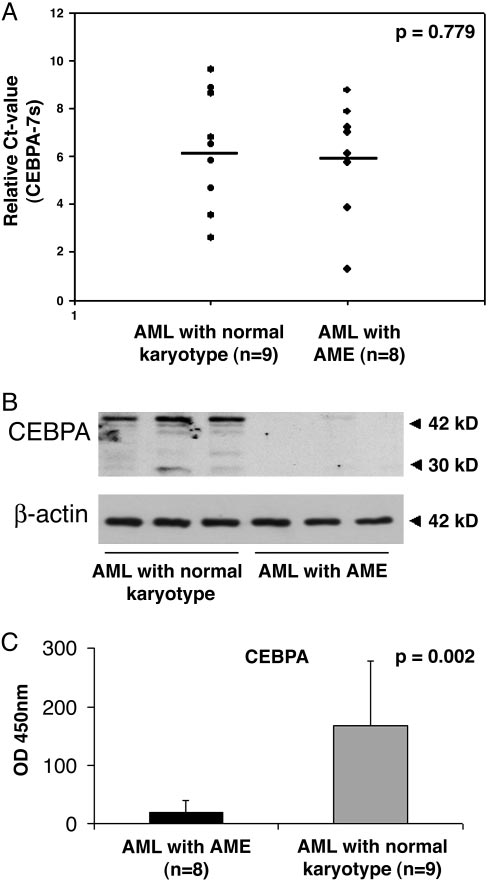
CEBPA Protein Is Specifically Suppressed in AML Patients Carrying the
effect on CEBPA mRNA and protein levels, parental U937 cells
AME Translocation. We aimed to verify the results obtained in
with the tetracycline-transactivator constructs but lacking the AME
induced U937 cells in malignant cells from eight patients carrying
cDNA were analyzed. Indeed, no changes on CEBPA mRNA and
the AME translocation. We compared them to nine AML patients
protein levels were detectable after withdrawal of tetracycline (data
with a normal karyotype. As assessed by direct sequencing, none of
the patients had CEBPA mutations. Real-time PCR analysis dem-
To investigate CEBPA DNA-binding activity, we performed
onstrated similar CEBPA mRNA levels (P ⫽ 0.779) in AML
gel-shift analyses. In unstimulated U937 cells, almost the entire
patients with the AME translocation and with a normal karyotype
binding activity to a CEBP site in a downstream target such as the
(Fig. 2A). Again, no CEBPA protein was detectable by Western blot
G-CSF receptor promoter is contributed by CEBPA (33). Starting
in any of the samples with the AME translocation. In contrast,
Helbling et al.

CRT Levels and Activity Are Increased After Conditional Expression of
AME in U937 Cells and in AML Patients with AME. There are only a few
reports about mechanisms involved in posttranscriptional regula-
tion of CEBPA (34–36). It has been shown that the poly(rC)-
binding protein hnRNP E2 inhibits CEBPA expression at the
translational level in patients with CML-BC but not in those in the
chronic phase of CML (CML-CP) (36). In contrast to patients with
CML-BC, we observed no changes of hnRNP E2 mRNA and
protein expression as assessed by real-time PCR and by Western
blot analysis in U937 cells following AME induction (data not
CRT has been reported to interact with CEBPA mRNA and
thereby to repress translation of the CEBPA protein (34). We
therefore hypothesized that the posttranscriptional down-
regulation of CEBPA after conditional expression of AME might
be caused by an increase of CRT expression and兾or activity.
Real-time PCR measurements of CRT mRNA transcripts showed
a 4.4-fold increase on day 2 (Fig. 3A). Western blot analysis of
whole-cell lysates further demonstrated an increase of CRT protein
after induction of AME (Fig. 3B).
CRT activity can be measured by UV cross-linking, thereby
enabling visualization of the direct interaction of CRT protein to a
CRT-binding site within the CEBPA mRNA (34). Fig. 3C gives
evidence of a dramatic increase in CRT activity starting early on day
1 after induction of AME (14.8-fold up-regulation). Finally, we
detected increased CRT activity in patient samples carrying the
AME translocation (12.2-fold) compared to AML patients with a
normal karyotype (Fig. 3D). In conclusion, results obtained from
patient samples and cell lines indicate that CEBPA-protein and
-binding activity seem to be regulated on a translational level by
modulation of CRT protein and activity.
We also tested in a single-cell assay whether overexpression of
CRT in U937 cells inhibits CEBPA translation in these cells. To
visualize cells expressing CRT, U937 cells were cotransfected with
CRT and with a vector expressing GFP at a 10:1 ratio. Under these
conditions, each green cell containing GFP is assumed to express
CRT (Fig. 3E). We observed that expression of CRT in U937 cells
inhibits translation of CEBPA. This CRT-dependent inhibition of
CEBPA translation is specific, because GFP alone does not affect
CEBPA expression. We examined the levels of CEBPA in 100 cells
transfected with CRT, and we found that CEBPA expression was
inhibited in 85 of the cells. Similar analysis of CEBPA protein in 100
cells transfected with GFP alone showed only nine cells with
reduced levels of CEBPA (Fig. 3E). Interestingly, no differences in
CEBPA protein is specifically suppressed in AME patients. Eight
patient samples carrying the AME translocation and nine AML patients with
CEBPA mRNA levels using real-time PCR were detected between
a normal karyotype were analyzed. (A) Real-time PCR analysis of CEBPA levels
U937 cells transfected with or without CRT (data not shown). Thus,
from AME patients and AML patients with a normal karyotype. Mean and SD
these studies indicate that overexpression of CRT blocks translation
(error bars) are shown. (B) Western blot analyses from lysates of three AME
of CEBPA in U937 cells.
patient samples and of three representative AML patients with a normal
karyotype. The same membrane was incubated with an antibody against
Inhibition of CRT by siRNA Restores CEBPA Protein Levels in AME U937
-actin for control (Lower). (C) CEBPA-binding activity was measured by using
Cells After Induction of AME. The experiments described above
the TransAM assay. Mean and SD (error bars) are shown.
suggest that CEBPA suppression is mediated by modulation of
CRT. We therefore hypothesized that functional knock-down of
significant amounts of CEBPA protein were seen in AML patients
CRT by siRNA might be able to restore efficient CEBPA transla-
with a normal karyotype (Fig. 2B).
tion. We thus induced the AME protein and transfected siRNA
We analyzed patient samples with and without the AME trans-
designed to target CRT. We observed an 87% knock-down of CRT
location for their binding activity to a CEBP consensus binding site
mRNA levels 48 h after transfection (Fig. 4A). Moreover, CRT-
by using the TransAM assay. We found a dramatically reduced
protein suppression was also evident 48 h after siRNA transfection
CEBPA-binding activity (92.1% reduction) in the eight samples
(Fig. 4B). The block of CRT-protein expression was equally ob-
with the AME translocation as compared to AML patients with a
served in U937 cells after AME induction as well as in the parental
normal karyotype (Fig. 2C). In contrast, no difference in binding
U937T cells.
activity was observed for CEBPB (P ⫽ 0.29) (data not shown).
Most interestingly, transfection of CRT siRNA prevented sup-
These results suggest that the leukemic fusion protein AME
pression of CEBPA protein after AME induction (Fig. 4B).
suppresses CEBPA protein and DNA-binding activity. In contrast
CEBPA-protein levels after inhibition of CRT by siRNA even
to our previous findings with the AML1-ETO fusion (32), no
exceeded the levels of U937 cells before AME induction (Fig. 4B).
changes on CEBPA mRNA levels were detected after induction of
In addition, the block of CRT in U937T cells (thus in the absence
AME. We therefore conclude that a posttranscriptional mechanism
of AME) also resulted in a significant increase of CEBPA protein.
must be involved in the regulation of CEBPA in AML with AME.
However, and in contrast to CEBPA, no significant changes in
Helbling et al.
PNAS 兩 September 7, 2004 兩 vol. 101 兩 no. 36 兩 13315

CRT expression and activity are induced after conditional expression of AME in U937 cells and AME patient samples. (A) Measurement of CRT mRNA
by real-time PCR. Mean and SD (error bars) are shown. (B) CRT proteins were assessed by Western blot analysis. HeLa cells served as positive control. (C) CRT activityof U937 AME cells after withdrawal of tetracycline was assessed by UV cross-linking. The assay demonstrates the direct interaction of CRT protein to a CRT-bindingsite within the CEBPA mRNA. Coomassie blue (C. blue) staining is given as a loading control. (D) CRT activity by UV cross-linking. Two representative AME patientsamples (lanes 1 and 2) are compared to two AML patient samples with a normal karyotype (lanes 3 and 4). Coomassie blue staining is depicted as a control. (E)U937 cells were transfected with GFP expression plasmid or GFP and CRT expression plasmids. Forty-eight hours after transfection, the expression of CEBPA wasexamined by immunostaining for CEBPA (Top). Levels of CEBPA expression were determined in 100 cells transfected with CRT and GFP and in 100 cells transfectedwith GFP alone (Bottom). DAPI, 4⬘,6-diamidino-2-phenylindole.
CEBPB expression were observed. We therefore conclude that
tiation in 32D cells and in murine bone marrow progenitors (17,
CRT in myeloid cells indeed is a potent inhibitor of CEBPA
37). Furthermore, it has been shown that AME inhibits the
antiproliferative effect of transforming growth factor . It also
blocks the granulocytic differentiation of IL-3-dependent 32D
cells when stimulated with G-CSF (16, 17). In addition, AME
Here we show that the myeloid transcription factor CEBPA is
seems to require functions and兾or functional cooperation of
specifically suppressed in AML patients carrying the AME trans-
both aml1 and evi1 to induce AML in mice (38). However, the
location. We also demonstrate that this suppression is mediated on
target genes involved in the differentiation block seen in AME
a translational level and is caused by CRT, a putative inhibitor of
leukemias remain unknown.
CEBPA translation (34).
In murine transplant models, AME can induce a disease similar
Previous reports by us and others have pointed to a crucial role
to human acute myelomonocytic leukemia (15). Coexpression of
of CEBPA in the pathogenesis of AML (29, 32, 36). In particular,
bcr-abl and ame fusion genes in mice rapidly induces AML,
the CBF complex seems to target CEBPA (32). Chromosomal
suggesting that a cooperation between mutations that dysregulate
abnormalities affecting AML1B兾RUNX1, one of the two subunits of
tyrosine kinase signaling (bcr-abl) and those that disrupt differen-
the transcription factor CBF, have been shown to suppress CEBPA
tiation (ame) is necessary (39). Interestingly, in CML-CP, CEBPA-
transcription (32). Here we focused on the AME translocation.
protein levels are normal, whereas patients with CML-BC have
Because the AME fusion equally affects the AML1 gene (the
suppressed CEBPA protein, thereby possibly contributing to the
DNA-binding subunit of the CBF complex), our findings further
differentiation block seen in CML-BC but not in CML-CP (36).
support the hypothesis that CBF leukemias target the myeloid key
Because AME does occur in patients with CML-BC, but not in
transcription factor CEBPA and that this pathway may contribute
CML-CP, one could speculate that the block in differentiation
to the differentiation block seen in these particular subsets of AML.
observed in patients with AME in CML-BC might be caused by
The mechanisms of how the AME fusion contributes to
CEBPA suppression. Moreover, a recent report suggests that
leukemogenesis are largely unknown. Expression of AME has
restoration of CEBPA in a BCR-ABL-positive cell line rapidly
been reported to increase proliferation and abnormal differen-
induces terminal granulocytic differentiation (28).
Helbling et al.

AML patients by dominant-negative mutations in the CEBPA gene
(29–31). We have further shown that CEBPA expression can be
inhibited by AML1-ETO on the transcriptional level by suppressing
its autoregulatory loop (32). Furthermore, a recent report indicates
that CEBPA expression in AML is abolished at the RNA level by
the tyrosine kinase receptor FLT3 (40). In addition, CEBPA
function seems to be inhibited by phosphorylation at Ser-21 me-
diated by overexpression of FLT3 or by FLT3 mutants (41).
Posttranscriptional modulation of CEBPA is involved in patients
with CML-BC through the inhibitory action of the poly(rC)-binding
protein hnRNP E2 by direct interaction with the upstream ORF of
CEBPA (36). A novel posttranscriptional mechanism for the mod-
ulation of CEBPA and CEBPB expression in HeLa cells was
reported recently, involving the chaperone CRT (34). CRT protein
binds to GCN repeats in the CEBPA mRNA and thereby impedes
translation of CEBPA mRNA. Our data suggest that in AME
leukemias, this mechanism is involved in the suppression of CEBPA
in vitro and in vivo, which highlights a role of RNA-binding proteins
for modulation of CEBPA expression in AML. It also suggests the
design of additional studies investigating the role of CRT in other
subsets of AML.
The myeloid key transcription factor CEBPA is believed to
suppress the leukemic phenotype through combined induction of
direct transcriptional targets crucial for normal myeloid differen-
tiation and inhibition of cell-cycle progression. We and others have
shown in leukemic cells that restoring CEBPA expression is suffi-
cient to induce neutrophil differentiation (25–29, 32, 40, 41),
thereby pointing to potential therapeutic implications. Here we
report that the block of CRT expression by siRNA powerfully
Inhibition of CRT expression by siRNA restores CEBPA-protein expres-
sion. AME protein was induced in U937 cells by withdrawal of tetracycline, and
restores CEBPA expression. Therefore, modulation of CRT ex-
siRNA designed to knock-down CRT or mock siRNA was transfected by elec-
pression might be a potent target for subsets of AML in which
troporation. As further control, U937T cells were transfected with siRNA. (A)
CEBPA protein is suppressed.
Real-time PCR analysis of CRT levels 48 h after transfection. Mean and SD (errorbars) are shown. (B) Western blot analysis of CRT, CEBPA, CEBPB, and -actin.
The AML1-MDS1-EVI1 cDNA was kindly provided by
Giuseppina Nucifora (Department of Pathology, University of Illinois,
Chicago), and the U937T cell line was obtained from Gerard Grosveld.
Various mechanisms have been reported thus far to account for
This work was supported by Swiss National Science Foundation Grants
a disruption of CEBPA function in AML patients. We and others
SF 31-666899.01 (to T.P.), SF 3100-67213 (to M.F.F.), and SF 3100A0-
have reported that wild-type CEBPA function is abrogated in some
100445 (to B.U.M.).
1. Tenen, D. G. (2003) Nat. Rev. Cancer 3, 89–101.
23. Swart, G. W., van Groningen, J. J., van Ruissen, F., Bergers, M. & Schalkwijk, J. (1997) Biol.
2. Rowley, J. D. (2001) Nat. Rev. Cancer 1, 245–250.
Chem. 378, 373–379.
3. Nucifora, G. (1997) Leukemia 11, 2022–2031.
24. Zhang, D. E., Zhang, P., Wang, N. D., Hetherington, C. J., Darlington, G. J. & Tenen, D. G.
4. Rubin, C. M., Larson, R. A., Anastasi, J., Winter, J. N., Thangavelu, M., Vardiman, J. W.,
(1997) Proc. Natl. Acad. Sci. USA 94, 569–574.
Rowley, J. D. & Le Beau, M. M. (1990) Blood 76, 2594–2598.
25. Radomska, H. S., Huettner, C. S., Zhang, P., Cheng, T., Scadden, D. T. & Tenen, D. G.
5. Rubin, C. M., Larson, R. A., Bitter, M. A., Carrino, J. J., Le Beau, M. M., Diaz, M. O. &
(1998) Mol. Cell. Biol. 18, 4301–4314.
Rowley, J. D. (1987) Blood 70, 1338–1342.
26. Wang, X., Scott, E., Sawyers, C. L. & Friedman, A. D. (1999) Blood 94, 560–571.
6. Nucifora, G., Begy, C. R., Kobayashi, H., Roulston, D., Claxton, D., Pedersen-Bjergaard, J.,
27. Miyamoto, T., Iwasaki, H., Reizis, B., Ye, M., Graf, T., Weissman, I. L. & Akashi, K. (2002)
Parganas, E., Ihle, J. N. & Rowley, J. D. (1994) Proc. Natl. Acad. Sci. USA 91, 4004–4008.
Dev. Cell 3, 137–147.
7. Osato, M., Asou, N., Abdalla, E., Hoshino, K., Yamasaki, H., Okubo, T., Suzushima, H.,
28. Tavor, S., Park, D. J., Gery, S., Vuong, P. T., Gombart, A. F. & Koeffler, H. P. (2003) J.
Takatsuki, K., Kanno, T., Shigesada, K. & Ito, Y. (1999) Blood 93, 1817–1824.
Biol. Chem. 278, 52651–52659.
8. Nucifora, G., Birn, D. J., Erickson, P., Gao, J., LeBeau, M. M., Drabkin, H. A. & Rowley,
29. Pabst, T., Mueller, B. U., Zhang, P., Radomska, H. S., Narravula, S., Schnittger, S., Behre,
J. D. (1993) Blood 81, 883–888.
G., Hiddemann, W. & Tenen, D. G. (2001) Nat. Genet. 27, 263–270.
9. Song, W. J., Sullivan, M. G., Legare, R. D., Hutchings, S., Tan, X., Kufrin, D., Ratajczak,
30. Preudhomme, C., Sagot, C., Boissel, N., Cayuela, J. M., Tigaud, I., de Botton, S., Thomas,
J., Resende, I. C., Haworth, C., Hock, R., et al. (1999) Nat. Genet. 23, 166–175.
X., Raffoux, E., Lamandin, C., Castaigne, S., Fenaux, P. & Dombret, H. (2002) Blood 100,
10. Roulston, D., Espinosa, R., 3rd, Nucifora, G., Larson, R. A., Le Beau, M. M. & Rowley, J. D.
(1998) Blood 92, 2879–2885.
31. Gombart, A. F., Hofmann, W. K., Kawano, S., Takeuchi, S., Krug, U., Kwok, S. H., Larsen,
11. Mikhail, F. M., Serry, K. A., Hatem, N., Mourad, Z. I., Farawela, H. M., El Kaffash, D. M.,
R. J., Asou, H., Miller, C. W., Hoelzer, D. & Koeffler, H. P. (2002) Blood 99, 1332–1340.
Coignet, L. & Nucifora, G. (2002) Cancer Genet. Cytogenet. 135, 96–100.
32. Pabst, T., Mueller, B. U., Harakawa, N., Schoch, C., Haferlach, T., Behre, G., Hiddemann,
12. Zent, C., Rowley, J. D. & Nucifora, G. (1997) Leukemia 11, Suppl. 3, 273–278.
W., Zhang, D. E. & Tenen, D. G. (2001) Nat. Med. 7, 444–451.
13. Mucenski, M. L., Taylor, B. A., Ihle, J. N., Hartley, J. W., Morse, H. C., 3rd, Jenkins, N. A.
33. Smith, L. T., Hohaus, S., Gonzalez, D. A., Dziennis, S. E. & Tenen, D. G. (1996) Blood 88,
& Copeland, N. G. (1988) Mol. Cell. Biol. 8, 301–308.
14. Morishita, K., Parker, D. S., Mucenski, M. L., Jenkins, N. A., Copeland, N. G. & Ihle, J. N.
34. Timchenko, L. T., Iakova, P., Welm, A. L., Cai, Z. J. & Timchenko, N. A. (2002) Mol. Cell.
(1988) Cell 54, 831–840.
Biol. 22, 7242–7257.
15. Cuenco, G. M., Nucifora, G. & Ren, R. (2000) Proc. Natl. Acad. Sci. USA 97, 1760–1765.
35. Lincoln, A. J., Monczak, Y., Williams, S. C. & Johnson, P. F. (1998) J. Biol. Chem. 273,
16. Sood, R., Talwar-Trikha, A., Chakrabarti, S. R. & Nucifora, G. (1999) Leukemia 13,
36. Perrotti, D., Cesi, V., Trotta, R., Guerzoni, C., Santilli, G., Campbell, K., Iervolino, A.,
17. Tanaka, T., Mitani, K., Kurokawa, M., Ogawa, S., Tanaka, K., Nishida, J., Yazaki, Y.,
Condorelli, F., Gambacorti-Passerini, C., Caligiuri, M. A., et al. (2002) Nat. Genet. 30, 48–58.
Shibata, Y. & Hirai, H. (1995) Mol. Cell. Biol. 15, 2383–2392.
37. Senyuk, V., Chakraborty, S., Mikhail, F. M., Zhao, R., Chi, Y. & Nucifora, G. (2002)
18. Scott, L. M., Civin, C. I., Rorth, P. & Friedman, A. D. (1992) Blood 80, 1725–1735.
Oncogene 21, 3232–3240.
19. Cao, Z., Umek, R. M. & McKnight, S. L. (1991) Genes Dev. 5, 1538–1552.
38. Cuenco, G. M. & Ren, R. (2004) Oncogene 23, 569–579.
20. Birkenmeier, E. H., Gwynn, B., Howard, S., Jerry, J., Gordon, J. I., Landschulz, W. H. &
39. Cuenco, G. M. & Ren, R. (2001) Oncogene 20, 8236–8248.
McKnight, S. L. (1989) Genes Dev. 3, 1146–1156.
40. Zheng, R., Friedman, A. D., Levis, M., Li, L., Weir, E. G. & Small, D. (2004) Blood 103,
21. Flodby, P., Barlow, C., Kylefjord, H., Ahrlund-Richter, L. & Xanthopoulos, K. G. (1996) J.
Biol. Chem. 271, 24753–24760.
41. Ross, S. E., Radomska, H. S., Wu, B., Zhang, P., Winnay, J. N., Bajnok, L., Wright, W. S.,
22. Chandrasekaran, C. & Gordon, J. I. (1993) Proc. Natl. Acad. Sci. USA 90, 8871–8875.
Schaufele, F., Tenen, D. G., & MacDougald, O. A. (2004) Mol. Cell. Biol. 24, 675–686.
Helbling et al.
PNAS 兩 September 7, 2004 兩 vol. 101 兩 no. 36 兩 13317
Source: http://international.insel.ch/fileadmin/innere-pupk/innere-pupk_users/Pdf/113761publication_2_PNAS.pdf
assets.yellow.co.nz
Making Education Easy Issue 6 - 2014 Welcome to the 6th issue of Fertility Research Review In this issue we review several studies investigating sperm quality. Many men are unaware that the use of finasteride, a common treatment for baldness, reduces sperm counts. Fortunately for couples desiring fertility, sperm counts were shown to increase on cessation of finasteride treatment. Couples may also
Icss protocol
The 2nd European Carotid Surgery Trial (ECST-2) Protocol Version 2.00 1 May 2013 International Standard Randomised Controlled Trial Number: ISRCTN97744893 ECST-2 Protocol v 2.0 – page 2 CONTENTS ECST-2 Protocol v 2.0 – page 3 PROTOCOL SUMMARY BACKGROUND Randomised trials have established the benefit of revascularisation by carotid endarterectomy (CEA) for moderate and severe carotid stenosis. However, a risk model derived from one of these trials and validated in another, showed that only patients with a high risk of stroke under medical therapy benefited from CEA. For a large range of patients there was neither clear benefit nor harm from CEA. Medical therapy for stroke prevention has improved since these original trials, with more widespread use of statins, more active lowering of blood pressure and more effective antiplatelet regimes. Lower optimum targets have been set for risk factor control e.g. blood pressure. Therefore CEA may not be beneficial in many patients with carotid stenosis treated by modern optimized medical therapy (OMT). HYPOTHESIS We hypothesize that in patients with carotid stenosis at low and intermediate risk for stroke, OMT alone is as effective in the long-term prevention of cerebral infarction and myocardial infarction (MI) as revascularisation and OMT combined. STUDY DESIGN ECST-2 is a multicentre, randomised, controlled, open, prospective clinical trial with blinded outcome assessment. We will use a risk model based on clinical characteristics to calculate a 5-year Carotid Artery Risk (mCAR) score, which will stratify patients as at high risk (≥15%), intermediate risk (7.5-15%), or low risk (<7.5%) of future stroke using predictive data from previous trials recalibrated to take account of the likely benefit of OMT. An interim analysis using MRI to determine the 2-year rates of cerebral infarction and haemorrhage after randomisation will be performed to assess safety and feasibility of the design and inform the design and sample size calculations for the full trial. ECST-2 will incorporate baseline imaging of carotid plaque where possible to investigate the predictive value of plaque characteristics. CENTRE REQUIREMENTS A neurologist or physician with an interest in stroke; a surgeon with expertise in CEA; if available, an interventionist with expertise in CAS. Access to MRI. INCLUSION CRITERIA Patients with symptomatic or asymptomatic atherosclerotic carotid artery stenosis (> 50%, NASCET criteria), suitable for revascularisation with CAR score indicating low or intermediate risk. MAIN EXCLUSION CRITERIA Patients with a CAR score indicating high risk, patients refusing either treatment, unable to consent or unsuitable for revascularisation due to anatomy, ill-health or disabling stroke (current Rankin >2). Recent contralateral carotid revascularisation, cardiac or other major surgery. RANDOMISATION AND TREATMENTS Patients will be randomly allocated in equal proportions to be treated by 1) immediate carotid revascularisation with OMT or 2) OMT alone (in the latter arm, revascularisation may be performed at a later stage if it becomes more clearly indicated e.g. because of TIA during follow up). Randomisation will be stratified by centre, type of planned revascularisation, symptom status and CAR score. A web-based randomisation system will be used. We anticipate that revascularisation will be by CEA in most patients, but carotid stenting (CAS) may be used if considered more appropriate. Centres will prespecify whether a patient will receive CEA or CAS if allocated to revascularisation. Randomisation and analysis will be stratified by the pre-specified intervention. The randomisation form will include entry of data to confirm a CAR score of <15%. OMT in both arms will consist of all three of: 1) optimal antiplatelet therapy; 2) statin or other cholesterol lowering treatment with target total cholesterol of <4 mmol/l and LDL cholesterol of <2 mmol/L; 3) antihypertensive treatment, if required, with target blood pressure of 135/85 mmHg. Patients will also undergo risk factor modification e.g. advice on smoking. FOLLOW UP The planned duration of follow up is a minimum of 5 years up to a maximum of 10 years. Recruitment and follow up will be supervised by the neurologist or stroke physician. Follow up will include ECG and troponin at 48 hours after revascularisation, with MRI at baseline and at 2 and 5 years follow up. SAMPLE SIZE The planned sample size is 320 patients for the safety MRI analysis and 2000 patients for the full trial. PRIMARY OUTCOME MEASURES For the full trial: any stroke at any time, plus non-stroke death occurring within 30 days of revascularisation. For the safety MRI analysis: The combined 2-year rate of cerebral infarction, cerebral haemorrhage, MI or periprocedural death after randomisation as assessed by follow up MRI and screening for MI. SECONDARY OUTCOME MEASURES Ipsilateral stroke, myocardial infarction, transient ischaemic attack or any hospitalisation for vascular disease during follow up. Disabling stroke during follow up. New cerebral infarction or haemorrhage on post procedural MRI. Ipsilateral restenosis or stenosis progression. Cognitive impairment. Further treatment procedure. Adverse events attributed to medical treatment or CEA. Quality of life and economic measures.